


What is cellular stress?
In an organ, there is a balance between the growth and death of the cells that make it up, which is called tissue homeostasis[1]. Each cell is able to grow to adapt to a physiological or pathological condition, change conformation or potentially multiply. It will also be capable of triggering a programmed death mechanism if the need arises or the pressure to which it is subjected is too great.
During cellular stress, this homeostasis is in danger and the cell will have several possibilities to respond depending on the type of stress. Overall, as long as the stimulus is manageable, the cell will adapt. Otherwise, it will have to put in place response mechanisms that may vary[2, 3].
The Heat Shock response
As the name suggests, this pathway was discovered by studying cells subjected to thermal cell stress[4]. It is now acknowledged that other stresses may activate it but the name has remained. This response consists in increasing the number and/or activity of chaperone proteins in order to prevent the proteins synthesized by the cell from folding incorrectly. The folding of proteins is essential to their proper function, and it is not uncommon for certain diseases to result from a protein synthesized correctly, but whose final conformation does not allow adequate functioning. In the case of cellular stress, proteins can lose their final shape, unfold, fold back or even aggregate with each other, when they should not. The heat shock response combats this effect, allowing the cell to continue to function in sub-optimal conditions. This response gives the cell a protection known as thermotolerance[5, 6, 7].

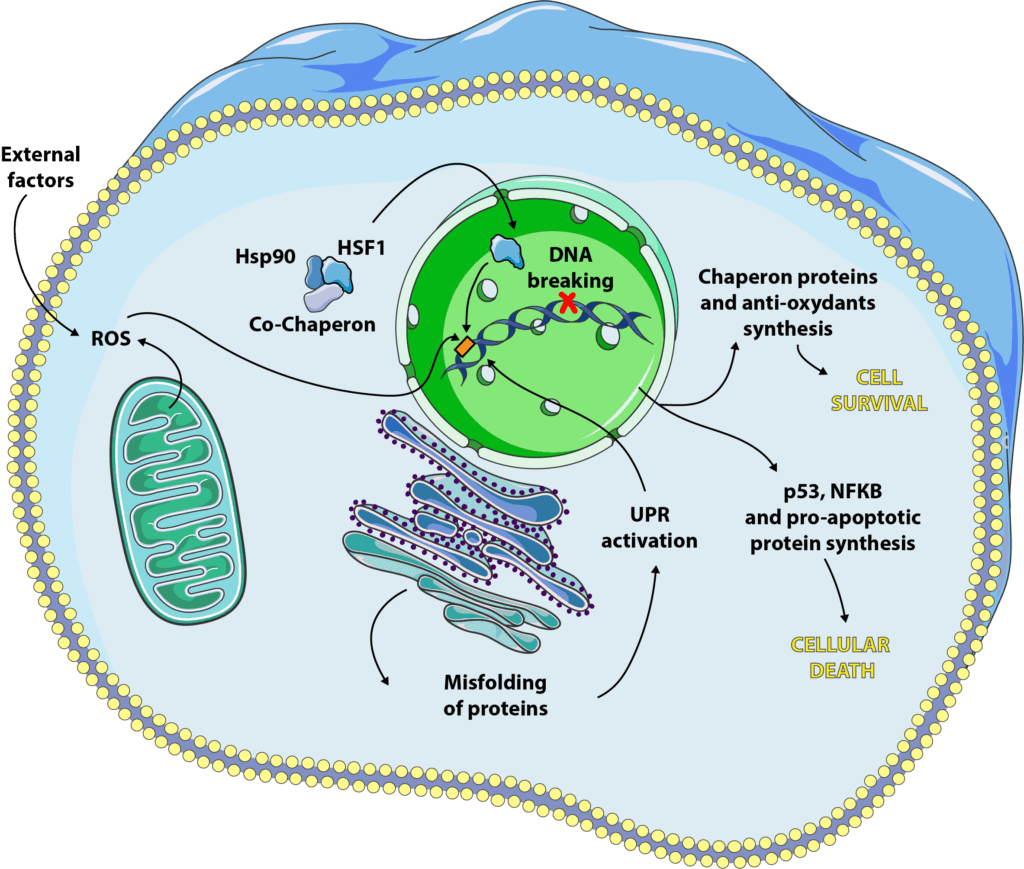
At first, gene transcription and translation is paused to avoid the synthesis of too many poorly folded proteins that the cell could not manage. In a second stage, transcription factors will come into play and activate the synthesis of specific molecules, heat shock factors (HSF)[8]. We have at our disposal 4 isoforms of these factors, HSF1, 2, 3 and 4 (yes, us scientists are not always very creative !), each having a different role, but of which the most studied for its function in the response to cellular stress is HSF1[9]. Under normal conditions, HSF1 keeps its inactive form, a cytoplasmic monomer, in interaction with chaperone proteins[10].
When the cell is subjected to stress, we observe an accumulation of poorly folded proteins that will compete with HSF1 to bind to chaperone proteins. From that moment on, HSF1 is released, will partner with its counterparts to form an active trimer that will bind to DNA and promote the specific expression of certain genes, including those encoding heat shock proteins (HSP).
HSP can be expressed constitutionally, as Hsp90 which is one of the chaperone proteins now HSF1 in its inactive form. Others will be specifically expressed during cellular stress, such as Hsp27 or Hsp70[5]. They will have different actions but will generally go in the direction of cellular survival. They can bind to poorly folded proteins[11] to prevent their aggregation, guarantee their function and maintain the integrity of the cytoskeleton[12]. They also block programmed cell death by inhibiting certain pro-apoptotic proteins such as caspases or cytochrome C[13, 14, 15].
The Unfolded Protein Response (UPR)
As we have seen previously, the correct folding of proteins is essential to their function, but for a protein to work properly, it must also undergo all kinds of post-translational modifications such as glycosylation, disulfide bridge formation, oligomerization… All this takes place in a specific organelle, the endoplasmic reticulum. Some shocks, such as caloric restriction or reduced oxygen supply, can cause endoplasmic reticulum stress, resulting in an influx of poorly folded, poorly modified proteins, a mechanism known as unfolded protein response (UPR)[16, 17]. This signalling pathway is activated via specific proteins which are, for the most part, present in inactive form under physiological conditions, and will be activated during cellular stress. These include IRE-1, PERK and ATF6, which will bind to DNA and encourage the synthesis of certain genes coding in particular for chaperone proteins and antioxidants[16].
The UPR is subjected to a delicate balance related to the ability of the reticulum to correctly fold the proteins and to manage those which are not. When UPR is able to improve this ability of the reticulum to cope with stress, via the synthesis of factors and proteins such as IRE-1, it will encourage cell survival. If the reticulum is overwhelmed, the UPR will rather promote cell death by mechanisms still little known.
The genotoxic response
The DNA contained in the nucleus of our cells is usually in double-stranded form, like a helix. During certain events, in particular DNA transcription and replication, it is found in single-stranded form: the enzymes that will allow these phenomena need access to the information contained in the DNA and will separate the two strands in order to copy or transcribe them. It is at this time that DNA is most sensitive to stresses such as radiation, ultraviolet radiation or anti-cancer treatments (sometimes called DNA intercalants). It will be possible to break the DNA either on one strand (single-strand break or SSB) or on both strands (double-strand break or DBS)[18]. Our cells are able to repair these breaks to some extent but sometimes they induce different signalling, particularly through p53 (a transcription factor that regulates apoptosis, autophagy and cell cycle) and NF-κB (a pro-inflammatory and pro-apoptotic cytokine).

Here again, everything is a matter of balance. When DNA damage is repairable, the signaling pathways engaged will be in favor of cell survival. If the breaks are too large, p53 and NF-κB come into play and will activate the cell death path.
These signalling routes are extremely complex, interconnected and still studied to this day.
Oxidative stress response
Reactive oxygen species (also called ROS) are one of the main causes of cellular stress. The survival of a cell depends on a balance between oxidants (oxygen and ROS) and antioxidants (enzymes and proteins such as glutathione, catalase or superoxide dismutases) that can be dysfunctional[19]. Most of the time, the cell is able to fight against oxidative stress and to promote its survival but, as for the previous cellular stresses mentioned, it happens that the oxidative:antioxidant balance is too deregulated for the cell to correct it. It will then enter processes of apoptosis or necrosis[19, 20, 21].
The best known ROS are hydrogen peroxide (H202, also called hydrogen peroxide), hydroxyl radicals (OH-) and nitric oxide (NO-). They have various origins, intra- or extracellular. The largest source of intracellular oxidants is our mitochondria and its respiratory chain: when it produces energy, it also produces ROS which are generally treated with superoxide dismutases. In addition to this intracellular source, our lifestyle greatly influences the generation of ROS. External causes include prolonged exposure to UV, pollution, pesticides, tobacco, alcohol, an unbalanced diet, too much sport, stress, or a nutritional deficiency in one or more antioxidants.
The response to oxidative stress combines a specific response and mechanisms common to other stresses, such as activation of chaperone protein synthesis[22] or NF-κB and p53[23]. It is not yet very clear how apoptosis induction takes place but it seems that H2O2 activates a system called Fas-FasL, which in turn induces caspase activation[24]. NO•, on the other hand, seems to be capable of inactivating certain antioxidant enzymes such as superoxide dismutase[25]. Interestingly, ROS could also activate a different cell death mechanism, necrosis. This occurs through the inactivation of caspases, normally pro-apoptotic molecules whose absence of stimulation will force the cell to turn to another form of death. The commitment to necrosis can also be explained by a decrease in ATP in the cell, inherent in damage to the mitochondria by too many oxidants[26]. Finally, a last mode can be engaged, that of autophagy, although the mechanisms leading to it are not yet clear[27].
Cellular stress and aging
Many of the stresses we have detailed worsen with age. Indeed, many diseases, in particular diabetes, neurodegenerative diseases (Parkinson and Alzheimer in the first line) or cancers, are partly caused by a deregulation of proteostasis and balance oxidants:antioxidants (see causes of aging). There is, for example, evidence that HSP synthesis increases with age[28] and is often coupled with deregulation of the expression of certain genes via the HSF1 signaling system, which functions less and less well[29]. Overall, stress, especially oxidative stress, is intimately linked to inflammation (via the production of cytokines, such as NF-κB) and when it becomes chronic, it is associated with an increase in ROS. It is a kind of loop that maintains itself and accelerates our aging. Cellular stress is therefore both a cause and a consequence of cellular pathologies and aging.
Some definitions
Cytokines: these are molecules synthesized by our immune cells that allow communication between cells. They are often associated with inflammation because they are the major actors in this process.
Chaperone proteins: these are small proteins whose role is to accompany the proteins during synthesis until they acquire their final conformation. They also prevent protein aggregation.
Transcription factor: it is a molecule that can bind to specific zones of DNA, called promoters, that regulate gene expression. They may have an activating or inhibiting role in transcription.
Monomer/dimer/protein trimer: when proteins form, to reach their final function, they sometimes need to bind together. When they leave the reticulum, they are in the form of monomer (i.e. a single peptide chain that forms a single protein). In the cytoplasm, they can then bind with one, two or more proteins forming dimers (two chains), trimers (three chains)… A distinction is made between homodimers, which are the combination of two identical proteins, and heterodimers, which result from the binding of two isoforms of the same protein or two different proteins.
Apoptosis: this is the mechanism triggered by the cell itself when it decides to die. Apoptosis is distinguished from necrosis by the signalling pathways involved in the establishment of the cell death process.
Autophagy: it is the mechanism by which a cell is able to get rid of certain dysfunctional organelles, by digesting them or by expelling them from its cytoplasm. When the cell manages to manage these dysfunctions, this way is pro-survival. On the other hand, when there are too many organelles that no longer work or when autophagy is not sufficient to treat them, this way will be pro-death.
All our articles on Cellular stress and aging:
Cellular stress and aging, a necessary duo?
 Although it is recognized as a risk factor by the scientific community, many questions remain regarding cellular stress. Its main role in aging-related diseases and in aging itself is still being studied.
Although it is recognized as a risk factor by the scientific community, many questions remain regarding cellular stress. Its main role in aging-related diseases and in aging itself is still being studied.
Part 1: The different types of cellular stress
 Cellular stress can mean many different mechanisms, whether the stress is mechanical, toxic, chemical, thermic, osmotic, ionizing… They all play a role in the aging process of our cells.
Cellular stress can mean many different mechanisms, whether the stress is mechanical, toxic, chemical, thermic, osmotic, ionizing… They all play a role in the aging process of our cells.
Part 2: The organelles targeted by cellular stress
 Cellular stress gets transmitted through several organelles in our cells, especially those of the mitochondria, the mitochondria and the nucleus, among others. Their response can vary and only give the cell under stress two viable options: adapt and survive, or die.
Cellular stress gets transmitted through several organelles in our cells, especially those of the mitochondria, the mitochondria and the nucleus, among others. Their response can vary and only give the cell under stress two viable options: adapt and survive, or die.
Part 3: Dealing with cellular stress
 In order to slow down the aging process of our cells, we must control our cellular stress. In this article, we will look into the techniques and/or treatments currently used to treat all forms of cellular stress.
In order to slow down the aging process of our cells, we must control our cellular stress. In this article, we will look into the techniques and/or treatments currently used to treat all forms of cellular stress.
References
[1] Simone Fulda, Adrienne M. Gorman, Osamu Hori, and Afshin Samali, “Cellular Stress Responses: Cell Survival and Cell Death,” International Journal of Cell Biology, vol. 2010, Article ID 214074, 23 pages.
[2]C. R. Weston and R. J. Davis, “The JNK signal transduction pathway,” Current Opinion in Cell Biology, vol. 19, no. 2, pp. 142–149, 2007.
[3] N. D. Perkins and T. D. Gilmore, “Good cop, bad cop: the different faces of NF-κB,” Cell Death and Differentiation, vol. 13, no. 5, pp. 759–772, 2006.
[4] S. Lindquist, “The heat-shock response,” Annual Review of Biochemistry, vol. 55, pp. 1151–1191, 1986.
[5] A. Samali and S. Orrenius, “Heat shock proteins: regulators of stress response and apoptosis,” Cell Stress and Chaperones, vol. 3, no. 4, pp. 228–236, 1998.
[6] A. Samali, C. I. Holmberg, L. Sistonen, and S. Orrenius, “Thermotolerance and cell death are distinct cellular responses to stress: dependence on heat shock proteins,” FEBS Letters, vol. 461, no. 3, pp. 306–310, 1999.
[7] G. C. Li and Z. Werb, “Correlation between synthesis of heat shock proteins and development of thermotolerance in Chinese hamster fibroblasts,” Proceedings of the National Academy of Sciences of the United States of America, vol. 79, no. 10, pp. 3218–3222, 1982.
[8] L. Pirkkala, P. Nykänen, and L. Sistonen, “Roles of the heat shock transcription factors in regulation of the heat shock response and beyond,” FASEB Journal, vol. 15, no. 7, pp. 1118–1131, 2001
[9] A. Shabtay and Z. Arad, “Reciprocal activation of HSF1 and HSF3 in brain and blood tissues: is redundancy developmentally related?” American Journal of Physiology, vol. 291, no. 3, pp. R566–R572, 2006.
[10] I. Shamovsky and E. Nudler, “New insights into the mechanism of heat shock response activation,” Cellular and Molecular Life Sciences, vol. 65, no. 6, pp. 855–861, 2008.
[11] F. U. Hartl and M. Hayer-Hartl, “Molecular chaperones in the cytosol: from nascent chain to folded protein,” Science, vol. 295, no. 5561, pp. 1852–1858, 2002.
[12] C. G. Concannon, A. M. Gorman, and A. Samali, “On the role of Hsp27 in regulating apoptosis,” Apoptosis, vol. 8, no. 1, pp. 61–70, 2003.
[13] J.-M. Bruey, C. Ducasse, P. Bonniaud et al., “Hsp27 negatively regulates cell death by interacting with cytochrome c,” Nature Cell Biology, vol. 2, no. 9, pp. 645–652, 2000.
[14] D. Chauhan, G. Li, T. Hideshima et al., “Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance,” Blood, vol. 102, no. 9, pp. 3379–3386, 2003.
[15] R. Steel, J. P. Doherty, K. Buzzard, N. Clemons, C. J. Hawkins, and R. L. Anderson, “Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1,” Journal of Biological Chemistry, vol. 279, no. 49, pp. 51490–51499, 2004.
[16] M. Schröder and R. J. Kaufman, “The mammalian unfolded protein response,” Annual Review of Biochemistry, vol. 74, pp. 739–789, 2005.
[17] D. Ron and P. Walter, “Signal integration in the endoplasmic reticulum unfolded protein response,” Nature Reviews Molecular Cell Biology, vol. 8, no. 7, pp. 519–529, 2007.
[18] M. Christmann, M. T. Tomicic, W. P. Roos, and B. Kaina, “Mechanisms of human DNA repair: an update,” Toxicology, vol. 193, no. 1-2, pp. 3–34, 2003.
[19] D. Trachootham, W. Lu, M. A. Ogasawara, N. R.-D. Valle, and P. Huang, “Redox regulation of cell survival,” Antioxidants & Redox Signaling, vol. 10, no. 8, pp. 1343–1374, 2008.
[20] K. Niizuma, H. Endo, and P. H. Chan, “Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival,” Journal of Neurochemistry, vol. 109, supplement 1, pp. 133–138, 2009.
[21] M. Genestra, “Oxyl radicals, redox-sensitive signalling cascades and antioxidants,” Cellular Signalling, vol. 19, no. 9, pp. 1807–1819, 2007.
[22] A. M. Gorman, B. Heavey, E. Creagh, T. G. Cotter, and A. Samali, “Antioxidant-mediated inhibition of the heat shock response leads to apoptosis,” FEBS Letters, vol. 445, no. 1, pp. 98–102, 1999.
[23] M. Meyer, R. Schreck, and P. A. Baeuerle, “H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor,” EMBO Journal, vol. 12, no. 5, pp. 2005–2015, 1993.
[24] T. L. Denning, H. Takaishi, S. E. Crowe, I. Boldogh, A. Jevnikar, and P. B. Ernst, “Oxidative stress induces the expression of Fas and Fas ligand and apoptosis in murine intestinal epithelial cells,” Free Radical Biology & Medicine, vol. 33, no. 12, pp. 1641–1650, 2002.
[25] M. Asahi, J. Fujii, K. Suzuki et al., “Inactivation of glutathione peroxidase by nitric oxide. Implication for cytotoxicity,” Journal of Biological Chemistry, vol. 270, no. 36, pp. 21035–21039, 1995.
[26] M. Leist, B. Single, H. Naumann et al., “Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis,” Experimental Cell Research, vol. 249, no. 2, pp. 396–403, 1999.
[27] R. Scherz-Shouval, E. Shvets, E. Fass, H. Shorer, L. Gil, and Z. Elazar, “Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4,” EMBO Journal, vol. 26, no. 7, pp. 1749–1760, 2007.
[28] Macario AJ, Conway de Macario E, Sick chaperones, cellular stress, and disease, N Engl J Med. 2005 Oct 6; 353(14):1489-501.
[29] Sóti C, Csermely P, Molecular chaperones and the aging process. Biogerontology. 2000; 1(3):225-33.
Dr. Marion Tible
Author/Reviewer
Auteure/Relectrice
Marion Tible has a PhD in cellular biology and physiopathology. Formerly a researcher in thematics varying from cardiology to neurodegenerative diseases, she is now part of Long Long Life team and is involved in scientific writing and anti-aging research.
More about the Long Long Life team
Marion Tible est docteur en biologie cellulaire et physiopathologie. Ancienne chercheuse dans des thématiques oscillant de la cardiologie aux maladies neurodégénératives, elle est aujourd’hui impliquée au sein de Long Long Life pour la rédaction scientifique et la recherche contre le vieillissement.
En savoir plus sur l’équipe de Long Long Life
Dr Guilhem Velvé Casquillas

Author/Reviewer
Auteur/Relecteur
Physics PhD, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
More about the Long Long Life team
Docteur en physique, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
En savoir plus sur l’équipe de Long Long Life











