


A review about longevity
Biological causes of aging and lifespan limitation. The more we learn about aging, the more it seems like it might not be an inescapable consequence of life. A striking fact is that aging rates vary dramatically from one species to the other, and from one individual to the other within one species. Some species age so slow that individuals systematically die of starvation, disease, accident or predation before showing any sign of senescence. It is thus tempting to imagine that a better knowledge of the determinants of lifespan could enable us to delay aging.

But could we reverse it?
Different perceptions of aging compete among scientists. Does aging follow a purposeful pattern driven by genes? Does it happen through random coincidental events? According to the evolution theory, all biological processes are genetically programmed from the beginning of life to reproductive maturity. Beyond this point, biologists are in disagreement as to whether genes further drive the organism into aging or whether aging stems from a stochastic accumulation of damage affecting molecular fidelity [1]. Aging thus defies a universally accepted definition.
Dr L. Hayflick
deplores that this leads to failures in communication and interpretation of research results [2]. He advocates a stochastic approach of aging and a deterministic view of longevity [2] [3]:

“Entropy explains aging, genetic determinism explains longevity, andundefined terminology explains misunderstanding both.”
L. Hayflick
The maintenance theory noticed that aging becomes manifest when molecular and cellular repair and replacement systems are no longer able to maintain the balance that existed before reproductive maturation. Some argue that this balance must ultimately fail because of increasing entropy [8].
Advocates of the disposable soma and the antagonistic pleiotropy theory see aging as a negative but inevitable side effect of necessary functions. Williams, who conceived the latter, wrote that natural selection could favor fitness in youth at the expense of fitness later on in life [9]:
“Natural selection may be said to be biased in favor of youth over old age whenever a conflict of interests arises.”
G. Williams
Other theories, suggesting that aging might be the result of a trade-off between some quality that produces a benefit in an organisms youth and disadvantage during old age, followed suit [10]. But why then does gene expression dramatically change during aging ? Inspired by the controversial group selection theory, scientists have shown that aging could benefit to survival of a group although compromising individual fitness. This point supports the programmed aging theories. As Wilson noticed in 1974 [11]:
“[To speak of programming] is to postulate that there are direct or indirect advantages of limiting life span through aging.”
Wilson
To learn more about this debate and its implications :
Aging can be tackled at different levels. First, there is a loss in molecular fidelity, so that some talk about molecular aging. Second, there is the aging of each cell, known as cell senescence. Cell senescence is a crucial mechanism for development but becomes deleterious when it affects stem and immune cells function, challenging tissue homeostasis. Aging is thus characterized by a changing phenotype at tissue and organ level. Recently, the hypothesis that aging could be driven by systemic factors has triggered intense scientific investigation.
This review examines the biological main causes of aging targeted by experimental anti-aging therapies. For complementary information, you can read The Hallmarks of Aging from Cell [12].

Genome damage, a limit to longevity?
DNA damage accumulates throughout life, because of the many factors that threaten genome stability. They include extrinsic physical, chemical, and biological agents, as well as intrinsic threats (DNA replication errors, hydrolytic reactions, and reactive oxygen species) [13]. Deficiencies in DNA repair mechanisms cause accelerated aging in mice and humans, suggesting causal links between accumulation of nuclear DNA damage and aging [13] [14] [15].
 Mitochondrial DNA damage. There is evidence of the implication of mitochondrial DNA (mtDNA) mutations in aging: deterioration of mtDNA through mutations [16] or defection of mtDNA polymerase [17] [18] [19] can induce premature aging or reduced lifespan. But whether mtDNA is more or less vulnerable to damage than nuclear DNA remains controversial. Besides, the multiple copies of mtDNA in each cell probably attenuate the consequences of mtDNA damage. Interestingly, the number of mitochondria decreases with age in liver cells of mice [20], rats [21], and humans [22] [23], consistent with a decrease in mtDNA copy number [24]. This could explain why mutation accumulation in mitochondrial genome might eventually become significant in the aging process. Another explanation is that accumulation of cells that contain high levels of mutant mtDNA could be an inevitable result of the normal mechanisms that maintain cellular concentrations of mtDNA [25].
Mitochondrial DNA damage. There is evidence of the implication of mitochondrial DNA (mtDNA) mutations in aging: deterioration of mtDNA through mutations [16] or defection of mtDNA polymerase [17] [18] [19] can induce premature aging or reduced lifespan. But whether mtDNA is more or less vulnerable to damage than nuclear DNA remains controversial. Besides, the multiple copies of mtDNA in each cell probably attenuate the consequences of mtDNA damage. Interestingly, the number of mitochondria decreases with age in liver cells of mice [20], rats [21], and humans [22] [23], consistent with a decrease in mtDNA copy number [24]. This could explain why mutation accumulation in mitochondrial genome might eventually become significant in the aging process. Another explanation is that accumulation of cells that contain high levels of mutant mtDNA could be an inevitable result of the normal mechanisms that maintain cellular concentrations of mtDNA [25].
Nuclear DNA damage. Somatic mutations and chromosomal anomalies are more likely to be observed in aged than young organisms [26] [27] [28]. DNA alterations eventually affect crucial genes or transcriptional pathways. As a result, cells dysfunction and may jeopardize tissue and organismal homeostasis, if not eliminated by apoptosis or senescence. Age-associated defects in the nuclear lamina may also aggravate genomic damage [29].
Recent studies suggest that nuclear DNA repair pathways might be involved in the aging process. The NAD+-dependent (1) poly(ADP-ribose) polymerase proteins (PARP) are usually associated with DNA repair mechanism. Recent studies have also linked them to mitochondrial function (4) and oxidative metabolism [30] [31]. PARP activation influences cellular metabolism through alterations in NAD+ (1) metabolism, direct PARylation events (a PARP-dependent post-translational alteration) and transcriptional reprogramming of the cell [31]. In general, PARP inhibition enhances oxidative metabolism and mitochondrial content (4). Interestingly, enhanced PARP activity is observed in aged organisms [32]. PARP-1 is an abundant nuclear enzyme activated by DNA damage and involved in repair mechanisms. Its activation may lead to rapid depletion of the cytosolic NAD+ (1) pool, and could even induce cell apoptosis [33] in case of massive DNA damage.
Another DNA damage response pathway is directed by tumor suppressor p53 (3). Interestingly, p53 can promote longevity by preventing the development of cancer, but increased p53 activation can have deleterious effects. It could promote aspects of the aging process by challenging tissue homeostasis with excessive senescence
or apoptosis signaling [34] ( see ”Cellular senescence” and ”Telomere shortening”).
As DNA damage accumulates with chronological age, the consumption of NAD+ (1) by PARP proteins increases. Sirtuins (2) activity is likely to be disturbed by NAD+ pool depletion. As for p53 (3), its overexpression in aging cells could be induced by PARP activation [35].
One interpretation is that DNA damage accumulation might be a biological clock driven by increasing entropy. DNA damage could be exponentially growing because it eventually affects genes encoding replication and repair mechanism, which thus become less efficient in faithfully replicating the DNA. With excessive activation of PARP, PARP-induced mechanism, designed to protect the organism against DNA damaged cell, may eventually deplete the NAD+ (1) pool and undermine NAD+-dependent sirtuins (2) function [36]. Although the role of sirtuins in the aging process is not fully unraveled yet, this alteration could have multiple consequences on cell metabolism.
Telomere shortening: Programmed cellular senescence?
DNA deterioration affects the genome almost randomly. But some chromosomal regions are particularly sensitive to aging-associated damage [37]. Replicative DNA polymerases do not replicate thoroughly the end of linear DNA molecules. This function is exclusively carried out by a special DNA polymerase known as telomerase. In humans, telomerase is only expressed in embryonic stem cells (enabling them to divide repeatedly), certain adult stem cells, and tumoral cells. Most somatic cells of mammals do not express telomerase. This leads to the progressive and cumulative loss of chromosome ends during cell cycles, and eventually entails cell senescence [38]. Telomerase deficiency in humans is associated with premature development of diseases (pulmonary fibrosis, dyskeratosis congenita, and aplastic anemia) which involve the loss of the regenerative capacity of different tissues [39].
 Several mechanisms are involved in regulating telomere replication. Shelterin is a protein complex that binds telomeres and regulates their replication [40] [41]. Loss-of-function models for shelterin components result in rapid decline of the regenerative capacity of tissues and accelerated aging [42]. Sirtuins are suspected to play a role in telomere maintenance [43] through epigenetic modulations. SIRT-6 modulates telomeric chromatin [44] and cell cycle [45].
Several mechanisms are involved in regulating telomere replication. Shelterin is a protein complex that binds telomeres and regulates their replication [40] [41]. Loss-of-function models for shelterin components result in rapid decline of the regenerative capacity of tissues and accelerated aging [42]. Sirtuins are suspected to play a role in telomere maintenance [43] through epigenetic modulations. SIRT-6 modulates telomeric chromatin [44] and cell cycle [45].
Critically short telomere length could activate a DNA damage response pathway involving p53 and p21WAF1 in aging cells [46]. p53 protein DNA binding and transcriptional activity increases with age, and p21WAF1 promoter activity in senescent cells is dependent on p53. Moreover, PARP could be involved in the post-translational activation of p53 in aging cells : p53 protein can associate with PARP, and inhibition of PARP activity leads to suppression of p21 expression in response to DNA damage [46]. PARP activation is associated with accelerated telomere loss and premature senescence [47].
Telomere shortening is often depicted as a biological clock for somatic cells, triggering cellular senescence. It looks like another anti-cancer strategy because it prevents cells from proliferating endlessly. This mechanism could thus promote early-life survival by reducing the development of cancer, but eventually challenge tissue homeostasis by triggering excessive senescence. Although it is sometimes interpreted as a programmed senescence mechanism (see ”Cellular senescence”), its role in phenotypic aging has not been clearly established.
Epigenetics as a key factor of lifespan control
Epigenetic mechanisms. Source : http://en.wikipedia.org/wiki/Epigenetics
Epigenetics is at the core of the aging process because it describes changes in cellular activity overtime through modulation of transcriptional pathways. Epigenetic alterations are induced by a complex enzymatic system ensuring the maintenance and the evolution of epigenetic patterns. They are associated with the activation of inflammatory genes and the development of age-related disease (cancer, dementia and atherosclerosis).
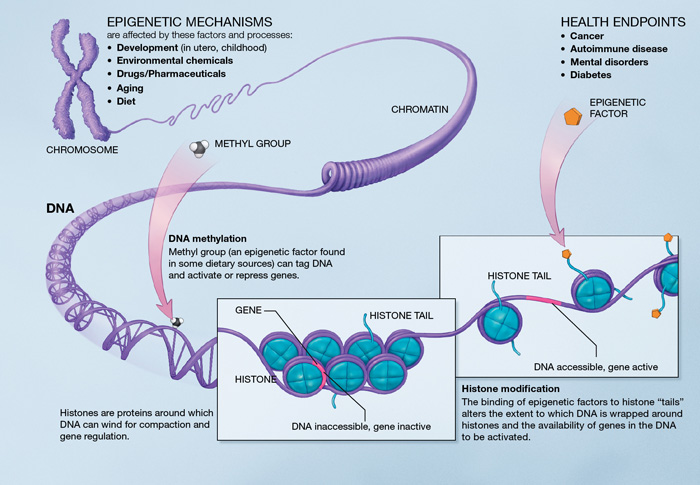
DNA methylation. There is an age-associated hypo- or hypermethylation of particular loci of the DNA [48]. On a gene promoter, methylation acts to repress gene transcription. Hypomethylation could increase DNA mutation rate. Hypermethylated genes with age include genes encoding estrogen receptor, insulin-like growth factor 2 (IGF-2), and tumor suppressor [48]. Remarkably, DNA methylation patterns modifications may particularly affect stem cells [49]. However, there is no experimental demonstration that lifespan can be extended by modifying DNA methylation patterns.
Chromatin remodeling. With age, there is a decrease in levels of chromatin remodeling factors [49] [50], as well as a loss or redistribution of global heterochromatin. Studies have found evidence of connection between heterochromatin formation and chromosomal stability [51]. Chromosome ends are assembled into heterochromatin domains [52] [53] so that epigenetic alterations can influence the regulation of telomere length.
Histone modifications. Deletion of components of histone methylation complexes extends longevity in invertebrates [54] [55]. Inhibition of histone demethylases in worms may extend lifespan by targeting components of key longevity routes (like IIS) [56]. Sirtuins SIRT-1, 6 and 7 (2) play a crucial role in histone modifications. In mammals, the benefits of SIRT-1 overexpression would include improved genomic stability [57] [58]. SIRT-6 regulates genomic stability, NF-κB signaling, and glucose homeostasis through histone deacetylation [59] [60] [61].
Transcriptional alterations. Age-associated increase in transcriptional noise [62] induces overproduction and maturation of mRNAs [63] [64] and miRNAs (see A systemic approach of aging – miRNAs). Transcriptional changes for genes encoding components of inflammatory, mitochondrial and lysosomal degradation pathways [65] show once again how intertwined the different hallmarks can be.
Aging and the deterioration of the unfolded protein response
Aging and many age-related diseases, in particular degenerative diseases (Alzheimer’s disease and age-related macular degeneration), have been associated with the cellular accumulation and aggregation of unfolded proteins [66], causing the cell to dysfunction. Protein homeostasis is balanced by complex intra-cellular mechanisms. In response to stress, those mechanisms are adapted to preserve functionality of the proteome.
Chaperone proteins play a key-role in protein folding, assembly, transport and targeting for degradation [67]. Age-associated changes challenge their activity and their synthesis [68] , inducing problems in protein quality control. Old organisms feature deteriorated ATP supply (see “Mitochondrial dysfunction”) that may interfere in chaperone function. Modifications in the substrate protein (for instance accumulation of advanced glycation end-products through non-enzymatic modifications [69]) can disrupt the chaperone’s ability to identify its target.
Pharmacological or genetic manipulations that increase lifespan in model organisms often stimulate autophagy (5) [70] [71]. Autophagy inhibition compromises the positive effects of CR, SIRT1 activation, inhibition of IIS, and administration of rapamycin, resveratrol, or spermidine [71] [72].
Several integrated pathways coregulate the degradation of proteins. Aging has a negative effect on the cross-talk between them [73] [74]. The aging-related deficiency of signaling pathways (insulin-like growth factor 1 (IGF-1) pathway, ensuring cross-talk between proteasome and autophagy (5)
The maintenance of mitochondrial proteostasis is challenging because of high levels of ROS species in this organelle. The most common way of eliminating dysfunctioning mitochondria may be autophagic degradation [75], known as mitophagy. However, numerous studies have reported dysfunctioning of mitochondrial clearance in aging and age-related disorders [76]. The mitochondrial Unfolded Protein Response (mtUPR) is activated when unfolded or misfolded proteins accumulate in the mitochondria [77] [78]. Studies show that communication between mitochondria and nucleus can induce the expression of nuclear genes encoding for mitochondrial proteins in response to impaired proteostasis [79] [80]. Mitochondria-nucleus communication failures could therefore be involved in the debilitation of mitochondrial proteostasis (see “Mitochondrial dysfunction”).
The mitochondrial sirtuin SIRT-3 (2), a protein deacylase, influences almost every major aspect of mitochondrial biology (including nutrient oxidation, ATP generation, ROS detoxification, mitochondrial dynamics, mtUPR) [81]. Proteotoxic stress induced by loss of mitochondrial proteostasis activates SIRT-3 and the antioxidant defense mechanisms of mitochondria [82]. The expression of SIRT-7, a component of the regulatory branch of the mtUPR, is reduced in aged hematopoietic stem cells, and its overexpression in this setting can improve the regenerative capability of these cells [83].
Mitochondria become dysfunctional with age
The oxidative stress theory (also known as free radical theory) argues that age-related mitochondrial dysfunction stems from high levels of reactive oxygen species (ROS) in the mitochondria (4) [84]. But the role of oxidative stress is likely to have been misunderstood during the early stages of research on aging. Recent experiments have shown that increased ROS levels may actually extend lifespan [85] [86] [87], whereas enhanced antioxidant defense did not [88]. Moreover, ROS have been found to spark off proliferation and survival responses in stress conditions [89]. The exact mechanism of ROS and oxidative stress-induced age-related pathologies is not yet very clear. They could for instance stimulate autophagy (5)
ROS could be regarded as a stress-induced survival signal. During aging, cellular stress and damage increase. The levels of ROS increase in parallel in an attempt to maintain survival. Beyond a certain point, ROS levels betray their homeostatic purpose, and eventually, they could aggravate age-associated damage [92].
 Mitonchondrial dysfunction seems to be a hallmark of aging. Remarkably, the mitochondrial OXPHOS metabolic pathway is disrupted during aging prior to significant mtDNA damage [93] : this suggests that mtDNA damage alone cannot account for mitochondrial dysfunction. Other causes have thus been studied. Mitochondria-nucleus communications are needed for mitochondria biogenesis and mitochondrial response to stress pathways [94]. Alterations of those communication pathways could trigger mitochondiral dysfunction.
Mitonchondrial dysfunction seems to be a hallmark of aging. Remarkably, the mitochondrial OXPHOS metabolic pathway is disrupted during aging prior to significant mtDNA damage [93] : this suggests that mtDNA damage alone cannot account for mitochondrial dysfunction. Other causes have thus been studied. Mitochondria-nucleus communications are needed for mitochondria biogenesis and mitochondrial response to stress pathways [94]. Alterations of those communication pathways could trigger mitochondiral dysfunction.
As chronological age advances, sirtuins (2) are deactivated. SIRT-1 and 3 downregulate HIF-1α (6) [93] [95] [96]. The downmodulation of HIF-1α induces production of mitochondrial transcription factor A (TFAM), a key activator of mitochondrial transcription. The depletion of the NAD+ pool (1) could then explain the deregulation of mitochondrial expression and OXPHOS disruption [93].
Sirtuins (2) are also involved in mitochondrial regulation through activation of PGC-1α(6) [97] [98]. Nucleotide excision DNA repair disorder triggers mitochondrial abnormalities that appear to be caused by decreased activation of the NAD/SIRT-1/PGC-1α axis through hyperactivation of the DNA damage sensor PARP-1 [99] (see “Genome damage”). p53 (3) expression can also repress PGC-1α and deregulate mitochondrial function, for instance as a result of telomere attrition [100].
Whether age-related mitochondrial dysfunction occurs through a PGC-1α– dependent, HIF-1α-dependent pathway, or a combination of both, is not clear yet.
Mitochondria (4) dysfunction could be induced by alteration of nuclear-mitochondria communication, possibly through mitochondrial sirtuins (2) activity deregulation. It has many negative effects on cell metabolism due to mitochondria’s crucial role in cell energetics and metabolism. For instance, it could provoke mismanagement of apoptosis and trigger inflammatory reactions [101].
Nutrient sensing: The benefits of calorie restriction for lifespan extention
Nutrient-sensing pathways adapt cell behaviour, metabolism, growth, development, and longevity to nutrient levels. One of them, the insulin/IGF-1 signaling (IIS) pathway is involved in glucose sensing and is linked to longevity. Hormone IGF-1 is produced in response to growth hormone (GH). Diverse genetic manipulations that attenuate signaling intensity at some point in the pathway (GH, IGF-1, AKT or FoxO) resulted in extended lifespan in worms, flies and mice. Multiple data suggest that calorie restrictions (CR)(7)
Other interconnected nutrient-sensing pathways include sirtuins (2), AMPK, and mTOR. AMPK and SIRT1 signal nutrient scarcity, whereas mTOR senses high amino acid concentrations. Sirtuins sense low energy states by detecting high NAD+ levels, and AMPK senses high energy states by detecting high AMP levels. Accordingly, their upregulation favors healthy aging. SIRT1 and AMPK responses are coordinated [106]. AMPK activation shuts off mTORC1 [107]. Metformin administration may extend lifespan through AMPK activation in worms and mice [108] [109] [110]. Rapamycin administration extends lifespan through mTOR inhibition [111]. As SIRT-1 activates PGC-1α (6) [98], PGC-1α could establish a connection between nutrient sensing and mitochondrial function.
Fasting also triggers lower PARP activity [112]. This reinforces the hypothesis that sirtuins (2) and PARP proteins “compete” for the cellular NAD+ pool (1).
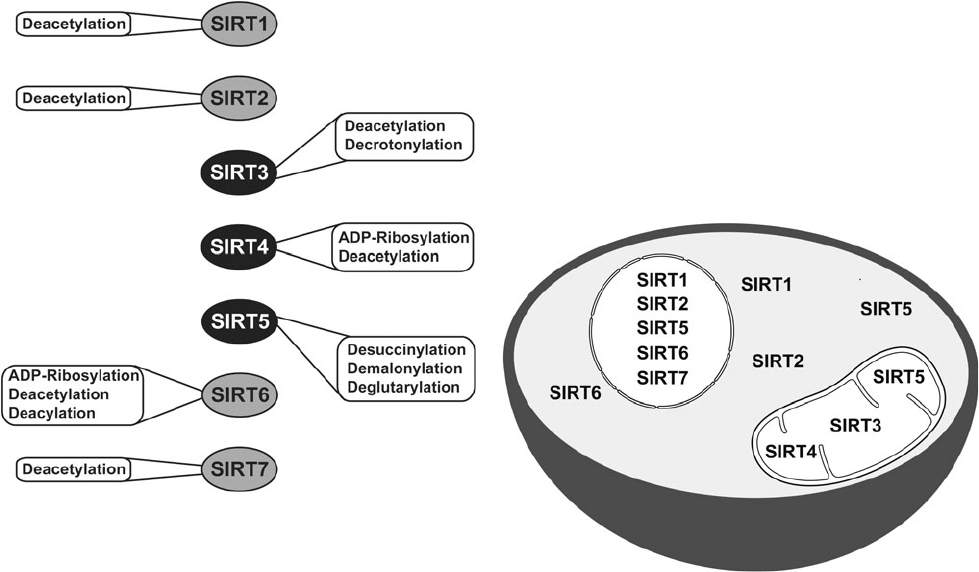
Catalytic activities and subcellular localization of mammalian sirtuins. Credit to David B. Lombard and Surinder Kumar, Mitochondrial sirtuins and their relationships with metabolic disease and cancer, 2015.
Paradoxically, low IIS is typical of normal aging phenotypes, whereas constitutively decreased IIS extends lifespan. A model in which decreased IIS is a defensive response to systemic damage can reconcile these results. Downmodulation of IIS is thought to entail slower metabolism and cell growth, and thus lower rate of cell damage. The deregulation of nutrient sensing can be interpreted as an organismal survival response, which ultimately becomes deleterious with age.
The principle of genetic determinism provides an appropriate underlying theory for research designs evaluating nutritional factors involved with lifespan. Nutritional studies are more accurately interpreted to have an impact on our understanding of longevity determination than aging [113].
Cellular senescence and phenotypic aging
A senescent cell stops dividing and features remarkable phenotypic changes, like the secretion of proinflammatory molecules [114]. Cell senescence is a crucial component of organismal development and is observed at every stage of life. It can be caused either by DNA damage (including telomere shortening), excessive mitogenic or oncogenic signaling, or the expression of a cell cycle inhibitor. Senescent cells accumulate in certain tissues with chronological aging [115]. Interestingly, senescence can be induced either by cellular endogenous pathways or by extra-cellular signaling pathways.
Two essential senescence pathways, activated in response to oncogenic alterations, have been identified : p16INK4a/Rb and p19ARF/p53 (3). The levels of tumor suppressor p16INK4a correlate with chronological age. p16INK4a and p19ARF are encoded by the same genetic locus (INK4a/ARF), which is associated with aging according to statistical studies.
Once again, there is disagreement over the effect of p16INK4a and p19ARF on lifespan. The activation of p53 (3) and INK4a/ARF could be regarded as a beneficial compensatory response to block the proliferation of damaged cells. However, when damage is pervasive, the regenerative capacity of tissues can be exhausted or saturated, and under these extreme conditions, the p53 and INK4a/ARF responses can accelerate aging.
Senescence might be a strategy to prevent a cell from proliferating into a tumoral cell. This anti-cancer system requires an efficient immune system to phagocyte senescent cells and robust stem cells to regenerate tissues. However, with chronological aging, senescent cells accumulate because of a slowdown of cell renewal and deteriorated immune mechanisms to clear senescent cells. Senescence is thus thought to promote early-life survival by preventing the development of cancer, but eventually to limit longevity as senescent cells accumulate. Deciding whether cellular senescence is a programmed aging mechanism needs further investigation : the link between cell senescence and phenotypic aging remains unclear.
Aging and exhaustion of stem cells
 Due to senescence of stem cells, aging tissues fail to regenerate as efficiently as younger tissues : they undergo fewer cell divisions. For instance, aging of hematopoietic stem cells entails a decrease in the production of adaptive immune cells and is thus involved in immunosenescence. Similar behaviours of stem cells are found in the brain, bone, and muscle fibers. DNA damage [116] and overexpression of proteins that inhibit cell cycle [117] could account for this behaviour.
Due to senescence of stem cells, aging tissues fail to regenerate as efficiently as younger tissues : they undergo fewer cell divisions. For instance, aging of hematopoietic stem cells entails a decrease in the production of adaptive immune cells and is thus involved in immunosenescence. Similar behaviours of stem cells are found in the brain, bone, and muscle fibers. DNA damage [116] and overexpression of proteins that inhibit cell cycle [117] could account for this behaviour.
Although adult stem cells express telomerase, they are not immune to telomere attrition [118] [119]. Accumulation of non-telomeric DNA damage and mutations has also been implicated in stem cell exhaustion [120] [121]. In the hematopoietic system, mutations that increase proliferation and survival are selected, and consistently, increased numbers of stem cells with pre-cancerous mutations are observed [122] [123] [124] [125]. DNA damage may also decrease stem cell numbers by causing them to undergo apoptosis or senescence.
Overexpression of sirtuin SIRT3 has been reported to enhance the regenerative capacity of old hematopoietic stem cells [126], suggesting sirtuins are also implicated in maintenance of stem cell function.
Stem cell exhaustion is suspected to play a prominent role in organismal aging, because it hampers cell renewal, an essential component of the aging process. Several hypotheses, described by Schultz and Sinclair in a 2016 review, try to explain how individual stem cells can function over the lifetime of an individual [127]. One of them is that stem cells use special mechanisms to prevent or reverse damage or epigenetic changes, including maintenance of long telomeres and enhanced proteostasis. Another hypothesis is that some stem cells decline with age while the healthiest are selected for. Models of improved stem cell function lead to improved tissue function consistently with what one would predict if stem cell aging is a crucial driver of tissue and organ aging [128] [129]. Understanding stem cell exhaustion has many implications for regenerative medicine.
A systemic approach of aging and lifespan determination
In 2014, by splicing animals together, scientists have shown that young blood can rejuvenate old tissues [130]. Moreover, lifespan-extending manipulations targeting one tissue can have beneficial consequences in other tissues [131] [132] [133]. These exciting results have triggered an active search for circulating systemic factors that could drive aging and coordinate the aging rate of different tissues and organs.
Age-related chronic inflammation
“Inflamm-aging” is a pro-inflammatory phenotype is observed in aging mammals [134]. Cytokines, the major players of the inflammatory response, allow communication between immune and somatic cells. Their chronic response might be partially responsible for frailty, development of age-related diseases, and functional decline [135] [136]. Multiple causes are examined : accumulation of proinflammatory tissue damage overtime, secretion of proinflammatory cytokines by senescent cells, dysfunctional immune system failing in efficiently clearing pathogens and dysfunctional host cells, mismanagement of apoptosis due to mitochondrial dysfunction, defective autophagy response and accumulation of damaged proteins triggering inflammatory response, change in the levels of hormones that regulate cytokine production (including testosterone), enhanced activation of NF-κB transcription factor.
These alterations result in an enhanced production of interleukin 1β (IL-1β), a tumor necrosis factor [101] [134] that induces the release of TNF-α. Interleukin-6 (IL-6) is released by TNF-α and IL-1β.
Nuclear factor-κB (NF-κB) is implicated in immune response and response to stress. It controls several genes associated with inflammation and is considered as a molecular culprit for inflammaging. The inhibition of NF-κB prevents age-associated features in different mouse models of accelerated aging [137] [138]. What is more, inhibiting NF-κB causes phenotypic rejuvenation of tissues [139]. Remarkably, SIRT-1 expression inhibits NF-κB.
Sirtuins could also influence inflammaging. Studies have revealed that, by deacetylating histones and components of inflammatory signaling pathways such as NF-κB, SIRT1 downregulates inflammation-related genes [140]. Reduction of SIRT-1 levels correlates with the development and progression of many inflammatory diseases, and pharmacological activation of SIRT1 may prevent inflammatory responses in mice [141] [142] [143]. SIRT-2 and 6 may downregulate the inflammatory response through deacetylation of NF-κB subunits and transcriptional repression of their target genes [60] [144].
Studies on the mRNA decay factor AUF-1 give in vivo evidence of the link between aging and inflammation. AUF1 directs cytokine mRNA degradation and participates in regulating the end of inflammatory response. AUF1-deficient mice feature a marked cellular senescence and premature aging phenotype. Interestingly, AUF1 also contributes to maintaining telomere length [145].
Once again, the age-associated inflammation can be interpreted as a defense mechanism that ultimately becomes deleterious overtime. Low levels of inflammatory response may be beneficial to tissue repair and regeneration by activating the immune system, but higher levels aggravate damage. Age-related inflammation could also severely undermine stem cell function [146].
![]()
miRNAs
Micro-RNAs (miRNAs) are a group of non-coding small RNAs. They can modulate gene expression by targeting mRNA with complementary sequence. Remarkably, miRNAs can modulate lifespan in invertebrates [147] [148] [149] by interacting with components of longevity networks or regulating stem cell regenerative abilities [150] [151] [152]. They could become an interesting biomarker of aging and age-related diseases, because miRNAs profiling reveals significant alterations in their expression levels [143].
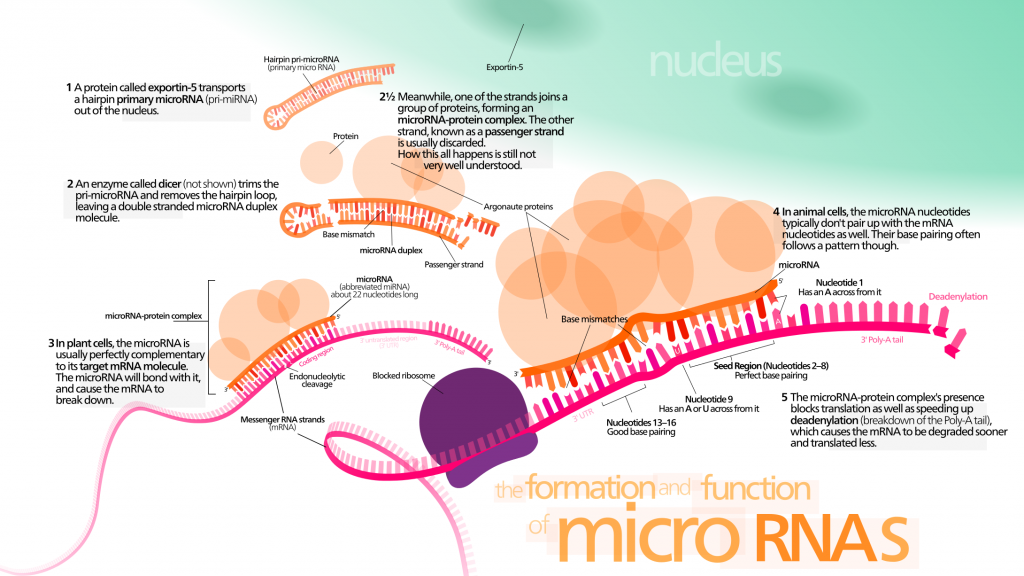
Cell-free miRNAs have raised biologists’ attention because they might provide useful information for early diagnosis of cancer and other age-related diseases. Circulating miRNAs could be major systemic factors of aging. They are known to be involved in several aspects of the aging process :
- Telomere length. miRNAs could be involved in the regulation of telomere length. Several miRNAs target mRNA implicated in telomere maintenance [153] ?? . Moreover, in humans, miRNA miR-138 is negatively correlated to telomerase reverse transcriptase (hTERT) levels [154].
- Senescence. There is evidence that miRNAs modulating the expression of p53 and p21 regulate cellular senescence [155]. SIRT-1 could deacetylate p53 and modulate its activity. miR-34a is a p53-induced miRNA, which can regulate the expression level of SIRT-1 in mice [156]. The connection of SIRT-1, p53, and miR-34a could hint at a senescence-regulating circuit. Moreover, in stress-induced and oncogene-induced senescence models, p21 is found to be the target of multiple miRNAs [157] [158]. Remarkably, levels of miR-106b family are decreased in senescent cells. In particular, miR-106a targets p21 mRNA [159] and prevents cell senescence whether p53 is inhibited or not. Other members of the miR-106b family enhance cell cycle by targeting p21 [158] [160]. miRNAs are suspected to stand at the crossroads between senescence and age-related diseases.
- Stem cell exhaustion. Some miRNAs regulate stem cell properties and could thus maintain cell-renewal capacity.
- Inflammation. miRNAs could be involved in inflammation regulation through interactions with NF-κB [161] [162]. A relatively small number of miRNAs seem to be involved in regulating inflammation : their prototypes are miR-155, miR-21, and miR-146a [163] [164]. In physiological conditions, their transcription is at baseline levels, but pro-inflammatory signaling results in strong co-induction of their expression through a NF-κB-dependent mechanism [165].
![]()
Thymus atrophy and its role in immuno-senescence
The thymus is an organ supporting the development and differentiation of T immune cells. T-cells are crucial to the adaptive immune system, which enables the body to respond specifically to foreign invaders.
In the 1980s, Steinman demonstrated that thymic function gradually decreases from the first year of life [166] and that the thymus is atrophied during aging [167]. This involution results in decreased efficiency of T-cell development, and reduced emigration of na¨ıve Tcells [168]. The adaptative immune system thus declines while the innate immune system is reinforced.
As the immune system becomes less efficient with age, the incidence of diseases such as opportunistic infections, autoimmunity and cancer increases [169] [170] increases. According to M. R. Dowling and P. D. Hodgkin, this apparently detrimental behavior may have been selected for to optimize fighting infections and avoid reaction to self [171]. Age-associated decline in immune function would thus be an unfortunate side effect of this selective process.
![]()
The role of the hypothalamus
The hypothalamus communicates with multiple tissues through hormonal and neural networks. It coordinates metabolic and behavioral responses to nutritional and environmental stimuli in mammals. It could thus be a crucial actor of systemic aging and of longevity control [172].
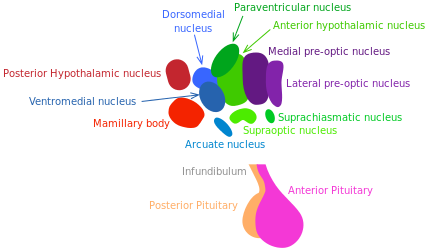
Growth hormone (GH) releasing hormone and somatostatin, respectively stimulating and inhibiting GH release, are produced by hypothalamic neurons. The GH/IGF-1 pathway (regulating growth, metabolism and tissue repair) is well established to control longevity in mammals [173] (see “Nutrient sensing pathways”).
Inflammatory responses activate NF-κB in the hypothalamus, inducing a signaling pathway that results in reduced production of GnRH by neurons. The decline of this hormone could enhance numerous aging-related systemic changes such as bone fragility, muscle weakness, skin atrophy, and reduced neurogenesis [174].
In 2013, Satoh et al. have demonstrated that brainspecific SIRT-1-overexpressing transgenic mice show lifespan extension. Excitingly, those mice exhibit phenotypes consistent with a delay in aging [175]. These phenotypes reveal enhanced neural activity, particularly in the dorsomedial and lateral hypothalamic nuclei. In this experiment, further information supported the longevity-promoting role of SIRT-1-mediated signaling in those areas of the hypothalamus. The findings of Satoh et al. suggest that SIRT-1 enhanced activity in the lateral and dorsomedail nuclei of the hypothalamus is crucial to in the systemic regulation of longevity in mammals.
![]()
Delphine Vendryes
Author
Auteure
Delphine is an engineer and was trained in bio-engineering and in biology at the École Polytechnique X in Paris.
More about the Long Long Life team
Delphine est désormais ingénieure, après une formation en biologie et bio-ingénierie à l’École Polytechnique X à Paris.
En savoir plus sur l’équipe de Long Long Life
Notes: Interesting components of aging pathways
(1) NAD
Nicotinamide adenine dinucleotide (NAD) is a coenzyme involved in many cell redox reactions, including ATP synthesis. It plays an essential role in many metabolic pathways, and is a substrate of both PARP proteins and sirtuins. Its oxidized form is known as NAD+, and its reduced form as NADH. NAD+ synthesis can occur either de novo from amino-acids, or by recycling preformed components. The cellular NAD+ pool could be depleted while aging [176]. This could be due to deterioration of synthesis pathways and/or enhanced degradation (possibly through PARP activation). NAD+ levels are thought to be a relevant molecular signature of aging. Precursors of NAD+ have been thoroughly studied because promoting the repletion of the NAD+ pool might activate sirtuins and help reverse aging.
(2) Sirtuins
Sirtuins are a family of NAD+-dependent proteins believed to be prominently implicated in the aging process. Sir2 was the first discovered sirtuin. In the 1990s, MIT Doctor Leonard Guarante found that sir2 gene controls longevity in yeast. Ortholog gene in C. Elegans proved crucial for longevity control too. Sirtuins are today thought to be key factors in longevity in many other species, including humans. Seven of them have been identified in mammals [177]. They have an enzymatic activity and can induce changes in cell metabolism and physiology. SIRT-1, 6 and 7 are nuclear proteins, they epigenetically influence gene expression by deacetylating histones [178]. SIRT-2 is involved in cell cycle control [178]. SIRT-3 and 5 regulate metabolic enzymes in the mitochondria [178]. They are believed to be involved in body response to calorie restriction.
(3) p53
p53 is a tumor suppressor known for its involvement in anti-cancer mechanism. It can activate DNA repair proteins and initiate apoptosis or senescence in response to DNA damage. Overexpression of p53 is deleterious because it drives excessive apoptosis and senescence [179]. p53 can either inhibit or stimulate autophagy depending on stress conditions [180] [181]. It might also affect aging and metabolism through its implication in mitochondrial function [182]. p53 protein DNA binding and transcriptional activity increases with age. p53 has an ambiguous role in the aging process, but it seems like it regulates an essential balance between aging and tumor suppression [183].
(4) Mitochondria
Mitochondria are cellular organelles that generate most of the cell’s adenosine triphosphate (ATP), used as a source of chemical energy. Oxidative phosphorylation (OXPHOS) is the metabolic pathway in which cells use enzymes to oxidize nutrients, releasing energy which is used to reform ATP. It takes place inside the mitochondria. Sirtuins are known to play a major role in mitochondrial activity regulation [178] [185]. Mitophagy is the deletion of mitochondria through autophagy. It happens as a response to damage or stress. Defective mitophagy (see ”Loss of proteostasis”) has been identified as a key factor for mitochondrial dysfunction [186].
(5) Autophagy
Autophagy is a regulated pathway responsible for the lysosomal degradation of cytoplasmic organelles or cytosolic components [181]. Autophagy activity decreases with age [72] [184], but is upregulated in response to calorie restrictions [71]. This upregulation can be interpreted as an effort to raise energy supplies when nutrients are scarce. Autophagy disruption is a key component of cellular aging, linked to mitochondrial dysfunction and loss of proteostasis.
(6) PGC-1α and HIF-1α
Activity of PGC-1α and HIF-1α can be modulated by sirtuins [187] [188]. Transcription factor PGC-1α directs a metabolic response that includes mitochondriogenesis, enhanced antioxidant defenses, and improved fatty acid oxidation [189]. HIF-1α is a subunit of transcription factor HIF-1. HIF-1 plays an important role in cellular response to systemic oxygen levels in mammals. It is involved in cell proliferation and survival and response to hypoxia [190]. HIF-1 also induces transcription of genes involved in cell proliferation and survival, as well as glucose and iron metabolism [191]. Gomes et al. have hypothesized that AMPK acts a switch between PGC-1α– dependent and HIF-1α-dependent regulation of mitochondria by sirtuins [93].
(7) Calorie restrictions
Calorie restrictions (CR) have been known to extend longevity in a variety of species, including mammals, since the 1930s. They are believed to favor mitochondrial oxidative metabolism and stress tolerance [177]. Studies tend to show that the lifespan-extending effect of CR depends on sirtuins [192] [193]. The evolution theory can bring an interesting insight into these mechanisms. When the energy supply in the environment is scarce, the metabolism switches to an energy-efficient mode. The aim is to ensure survival to be alive when the environment becomes more favorable to reproduction.
![]()
References
- Finch, C.E.; Kirkwood, T.B. Chance, Development, and Aging; Oxford University Press: New York, NY, USA, 2000.
- Hayflick, L. (2007). Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS genet, 3(12), e220.
- Hayflick, L. Biological aging is no longer an unsolved problem. Ann. N. Y. Acad. Sci. 2007, 1100, 113.
- Hamilton, W.D. The moulding of senescence by natural selection. Theor. Biol. 1966 , 12, 1245.
- Rose, M.R.; Burke, M.K.; Shahrestani, P.; Mueller, L.D. Evolution of ageing since Darwin. Genet. 2008, 87, 363371.
- Lithgow, G.J. Why aging isnt regulated: A lamentation on the use of language in aging literature. Gerontol. 2006, 41, 890893.
- Medawar, P. B. (1952). An unsolved problem of biology.
- Lambert F.L. (2007). Entropy and the second law of thermodynamics.
- Williams, G.C. (1957) Pleiotropy, natural selection and the evolution of senescence. Evolution11, 398411.
- Kirkwood, T. B. (2002). Evolution of ageing. Mechanisms of ageing and development, 123(7), 737-745.
- Wilson, D. L. (1974). The programmed theory of aging. Theoretical aspects of aging, 11-21.
- Lo´pez-Ot´ın, C., Blasco, M. A., Partridge, L., Serrano, M., and G. Kroemer, G. (2013). The Hallmarks of Aging, Cell 153
- Hoeijmakers, J.H. (2009). DNA damage, aging, and cancer. Engl. J. Med. 361 , 14751485
- Gregg, S.Q., Guti´errez, V., Robinson, A.R., Woodell, T., Nakao, A., Ross, M.A., Michalopoulos, G.K., Rigatti, L., Rothermel, C.E., Kamileri, I., et al. (2012). A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology 55 , 609621.
- Murga, M., Bunting, S., Montan˜a, M.F., Soria, R., Mulero, F., Can˜amero, M., Lee, Y., McKinnon, P.J., Nussenzweig, A., and Fernandez-Capetillo, O. (2009). A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Genet. 41, 891898.
- Wallace, D.C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Rev. Genet. 39, 359407.
- Kujoth, G.C., Hiona, A., Pugh, T.D., Someya, S., Panzer, K., Wohlgemuth, S.E., Hofer, T., Seo, A.Y., Sullivan, R., Jobling, W.A., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481484.
- Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J.N., Rovio, A.T., Bruder, C.E., Bohlooly-Y, M., Gidlo¨f, S., Oldfors, A., Wibom, R., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417423.
- Vermulst, M., Wanagat, J., Kujoth, G.C., Bielas, J.H., Rabinovitch, P.S., Prolla, T.A., and Loeb, L.A. (2008). DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Genet. 40, 392394.
- Herbener G. A morphometric study of age-dependent changes in mitochondrial populations of mouse liver and heart. J Gerontol. 1976;31(1):812.
- Stocco DM, Hutson JC. Quantitation of mitochondrial DNA and protein in the liver of Fischer 344 rats during aging. J Gerontol. 1978;33(6):802809.
- Yen T-C, Chen Y-S, King K-L, Yeh S-H, Wei Y-H. Liver mitochondrial respiratory functions decline with age.
- Tauchi H, Sato T. Age changes in size and number of mitochondria of human hepatic cells. J Gerontol. 1968;23(4):454461.
- Stocco DM, Cascarano J, Wilson MA. Quantitation of mitochondrial DNA, RNA, and protein in starved and starved-refed rat liver. J Cell Physiol. 1977;90(2):295306.
- Chinnery, P. F., Samuels, D. C., Elson, J., Turnbull, D. M. (2002). Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism?. The Lancet, 360(9342), 1323-1325.
- Moskalev, A.A., Shaposhnikov, M.V., Plyusnina, E.N., Zhavoronkov, A., Budovsky, A., Yanai, H., and Fraifeld, V.E. (2012). The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev.
- Faggioli, F., Wang, T., Vijg, J., and Montagna, C. (2012). Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Mol. Genet. 21, 52465253.
- Forsberg, L.A., Rasi, C., Razzaghian, H.R., Pakalapati, G., Waite, L., Thilbeault, K.S., Ronowicz, A., Wineinger, N.E., Tiwari, H.K., Boomsma, D., et al. (2012). Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 90, 217228.
- Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shumaker, D.K., Solimando, L., and Goldman, R.D. (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832853.
- Mouchiroud, L., Houtkooper, R.H., Moullan, N., Katsyuba, E., Ryu, D., Canto´, C., Mottis, A., Jo, Y.S., Viswanathan, M., Schoonjans, K., Guarente, L., Auwerx, J. (2013) The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Jul 18;154(2):430-41. doi: 10.1016 /j.cell. 2013.06.016.
- Bai, P., Cant´o, C. (2012). The Role of PARP-1 and PARP-2 Enzymes in Metabolic Regulation and Disease Cell Metabolism 16, September 5
- Braidy, N., Guillemin, G.J., Mansour, H., Chan-Ling, T., Poljak, A., and Grant, R. (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 6, e19194.
- Ying, W., Alano, C.C., Garnier, P., Swanson R.A. (2005). NAD as a Metabolic Link Between DNA Damage and Cell Death. J Neurosci Res. Jan 1-15;79(1-2):216-23
- Campisi, J. (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell, 120(4), 513-522.
- Agarwal, M. L., Taylor, W. R., Chernov, M. V., Chernova, O. B., Stark, G. R. (1998). The p53 network. Journal of Biological Chemistry, 273(1), 1-4.
- Guarente, L. (2014). Linking DNA Damage, NAD+/SIRT1, and Aging. Cell Metabolism 20, November 4.
- Blackburn, E.H., Greider, C.W., and Szostak, J.W. (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Med. 12, 11331138.
- Shay, J. W., Wright, W. E. (2005). Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis, 26(5), 867-874.
- Armanios, M., and Blackburn, E.H. (2012). The telomere syndromes. Nat. Rev. Genet. 13, 693704.
- Palm, W., de Lange, T. (2008). How shelterin protects mammalian telomeres. Rev. Genet. 42, 301334.
- de Lange T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19: 2100-2110.
- Mart´ınez, P., and Blasco, M.A. (2010). Role of shelterin in cancer and aging. Aging Cell 9, 653666.
- Jia, G., Su, L., Singhal, S., Liu, X. (2012). Emerging roles of SIRT6 on telomere maintenance, DNA repair, metabolism and mammalian aging. Mol Cell Biochem. May;364(1-2):345-50. doi: 10.1007/s11010-012-1236-8
- Michishita, E., McCord, R.A., Elisabeth Berber, E., Mitomu Kioi, M., Padilla-Nash, H., Damian, M., Cheung, P., Kusumoto, R., Kawahara, T.L.A., Barrett, J.C., Chang, H.Y., Bohr, V.A., Ried, T., Gozani, O., Chua, K.F. (2008). SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. Mar 27;452(7186):492-6.
- Michishita, E., McCord, R.A., Boxer, L.D., Barber, M.F., Hong, T., Gozani, O., Chua, K.F. (2009). Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6.
- Herbig, U., Jobling, W. A., Chen, B. P., Chen, D. J., Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21 CIP1, but not p16 INK4a. Molecular cell, 14(4), 501-513.
- Vaziri, H., West, M.D., Allsopp, R.C., Davison, T.S., Wu, Y.S., Arrowsmith, C.H., Poirier, G.G., Benchimol, S. (1997). ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. Oct 1;16(19):6018-33.
- Maegawa, S., Hinkal, G., Kim, H.S., Shen, L., Zhang, L., Zhang, J., Zhang, N., Liang, S., Donehower, L.A., and Issa, J.P. (2010). Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 20, 332340.
- Pollina, E.A., and Brunet, A. (2011). Epigenetic regulation of aging stem cells. Oncogene 30, 31053126.
- Pegoraro, G., Kubben, N., Wickert, U., Go¨hler, H., Hoffmann, K., and Misteli, T. (2009). Ageing-related chromatin defects through loss of the NURD complex. Cell Biol. 11, 12611267.
- Schotta, G., Lachner, M., Sarma, K., Ebert, A., Sengupta, R., Reuter, G., Reinberg, D., and Jenuwein, T. (2004). A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 12511262.
- Blasco, M.A. (2007). The epigenetic regulation of mammalian telomeres. Rev. Genet. 8, 299309.
- Gonzalo, S., Jaco, I., Fraga, M.F., Chen, T., Li, E., Esteller, M., and Blasco, M.A. (2006). DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Cell Biol. 8, 416424.
- Greer, E.L., Maures, T.J., Hauswirth, A.G., Green, E.M., Leeman, D.S., Maro, G.S., Han, S., Banko, M.R., Gozani, O., and Brunet, A. (2010). Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383387.
- Siebold, A.P., Banerjee, R., Tie, F., Kiss, D.L., Moskowitz, J., and Harte, P.J. (2010). Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Natl. Acad. Sci. USA 107, 169174.
- Jin, C., Li, J., Green, C.D., Yu, X., Tang, X., Han, D., Xian, B., Wang, D., Huang, X., Cao, X., et al. (2011). Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 14, 161172.
- Oberdoerffer, P., Michan, S., McVay, M., Mostoslavsky, R., Vann, J., Park, S.K., Hartlerode, A., Stegmuller, J., Hafner, A., Loerch, P., et al. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907918.
- Wang, R.H., Sengupta, K., Li, C., Kim, H.S., Cao, L., Xiao, C., Kim, S., Xu, X., Zheng, Y., Chilton, B., et al. (2008). Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312323.
- Kanfi, Y., Peshti, V., Gil, R., Naiman, S., Nahum, L., Levin, E., Kronfeld-Schor, N., and Cohen, H.Y. (2010). SIRT6 protects against pathological damage caused by dietinduced obesity. Aging Cell 9, 162173.
- Kawahara, T.L., Michishita, E., Adler, A.S., Damian, M., Berber, E., Lin, M., McCord, R.A., Ongaigui, K.C., Boxer, L.D., Chang, H.Y., and Chua, K.F. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 6274.
- Zhong, L., DUrso, A., Toiber, D., Sebastian, C., Henry, R.E., Vadysirisack, D.D., Guimaraes, A., Marinelli, B., Wikstrom, J.D., Nir, T., et al. (2010). The histone deacetylase Sirt6 regulates glucose homeostasis via HIF1α. Cell 140, 280293.
- Bahar, R., Hartmann, C.H., Rodriguez, K.A., Denny, A.D., Busuttil, R.A., Doll, M.E., Calder, R.B., Chisholm, G.B., Pollock, B.H., Klein, C.A., and Vijg, J. (2006). Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441, 10111014.
- Harries, L.W., Hernandez, D., Henley, W., Wood, A.R., Holly, A.C., BradleySmith, R.M., Yaghootkar, H., Dutta, A., Murray, A., Frayling, T.M., et al. (2011). Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 10, 868878.
- Nicholas, A., de Magalhaes, J.P., Kraytsberg, Y., Richfield, E.K., Levanon, E.Y., and Khrapko, K. (2010). Age-related gene-specific changes of A-to-I mRNA editing in the human brain. Ageing Dev. 131, 445447.
- de Magalha˜es, J.P., Curado, J., and Church, G.M. (2009). Meta-analysis of agerelated gene expression profiles identifies common signatures of aging. Bioinformatics 25, 875881.
- Kaushik, S., Cuervo, A.M. (2015). Proteostasis and aging. Nature Medicine 21, 14061415
- Feldman, D.E., Frydman, J. Protein folding in vivo: the importance of molecular chaperones. Opin. Struct. Biol. 10, 2633 (2000).
- Calderwood, S.K., Murshid, A., and Prince, T. (2009). The shock of aging: molecular chaperones and the heat shock response in longevity and aginga mini-review. Gerontology 55, 550558.
- Vanhooren, V., Navarrete Santos, A., Voutetakis, K., Petropoulos, I., Libert, C., Simm, A., Gonos, E.S., Friguet, B. (2015). Protein modification and maintenance systems as biomarkers of ageing. Mech Ageing Dev. Nov;151:71-84.
- Morselli, E., Galluzzi, L., Kepp, O., Criollo, A., Maiuri, M. C., Tavernarakis, N., … Kroemer, G. (2009). Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging, 1(12), 961-970.
- Morselli, E., Maiuri, M.C., Markai, M., Megalou, E., Pasparaki, A., Paliaras, K., Galluzzi, L., Criollo, A., Malik, S.A., Madeo, F., et al. (2010). Caloric restriction and resveratrol prolong longevity via the sirtuin-1 dependent induction of autophagy. Cell Death Disease 1, e10.
- Rubinsztein, D.C., Marino, G., Kroemer, G. (2011) Autophagy and aging. Cell 146 , 682695.
- Gavil´an, E., Pintado, C., Gavilan, M.P., Daza, P., S´anchez-Aguayo, I., Castan˜o, A., Ruano, D. (2015). Age-related dysfunctions of the autophagy lysosomal pathway in hippocampal pyramidal neurons under proteasome stress. Aging 36, 19531963.
- Schneider, J.L., Villarroya, J., Diaz-Carretero, A., Patel, B., Urbanska, A.M., Thi, M.M., Villarroya, F., Santambrogio, L., Cuervo, A.M. (2015). Loss of hepatic chaperonemediated autophagy accelerates proteostasis failure in aging. Aging Cell 14, 249264.
- Lemasters, J.J. (2014). Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2, 749754.
- Jensen, M.B., Jasper, H. (2014). Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 20, 214225.
- Heo, J.M., Livnat-Levanon, N., Taylor, E.B., Jones, K.T., Dephoure, N., Ring, J., Xie, J., Brodsky, J.L., Madeo, F., Gygi, S.P., Ashrafi, K., Glickman, M.H., Rutter, J. (2010). A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465480.
- Braun, R.J., Sommer, C., Leibiger, C., Gentier, R.J., Dumit, V.I., Paduch, K., Eisenberg, T., Habernig, L., Trausinger, G., Magnes, C., Pieber, T., Sinner, F., Dengjel, J., van Leeuwen, F.W., Kroemer, G., Madeo, F. (2015). Accumulation of basic amino acids at mitochondria dictates the cytotoxicity of aberrant ubiquitin. Cell Rep. 10, 15571571.
- Jovaisaite, V., Auwerx, J. (2015). The mitochondrial unfolded protein response synchronizing genomes. Curr. Opin. Cell Biol. 33, 7481.
- Haynes, C.M., Ron, D. (2010). The mitochondrial UPR – protecting organelle protein homeostasis. Cell Sci. 123, 38493855.
- McDonnell, E., Peterson, B.S., Bomze, H.M., Hirschey, M.D. (2015). SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. 26, 486492.
- Papa, L., Germain, D. (2014). SirT3 Regulates the Mitochondrial Unfolded Protein Response. Mol Cell Biol. Feb;34(4):699-710.
- Mohrin M., Shin J., Liu Y., Brown K., Luo H., Xi Y., Haynes C.M., Chen D. (2015). Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 13741377.
- Harman, D. (1965). The free radical theory of aging : effect of age on serum copper levels, J. Gerontol 20, 151-153
- Doonan, R., McElwee, J.J., Matthijssens, F., Walker, G.A., Houthoofd, K., Back, P., Matscheski, A., Vanfleteren, J.R., and Gems, D. (2008). Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 22, 32363241.
- Mesquita, A., Weinberger, M., Silva, A., Sampaio-Marques, B., Almeida, B., Le˜ao, C., Costa, V., Rodrigues, F., Burhans, W.C., and Ludovico, P. (2010). Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Natl. Acad. Sci. USA 107, 1512315128.
- Van Raamsdonk, J.M., and Hekimi, S. (2009). Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000361
- Prez, V.I., Bokov, A., Van Remmen, H., Mele, J., Ran, Q., Ikeno, Y., Richardson, A., (2009). Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta, 1790, pp. 10051014
- Sena, L.A., and Chandel, N.S. (2012). Physiological roles of mitochondrial reactive oxygen species. Cell 48, 158167.
- Bellot, G. L., Liu, D., Pervaiz, S. (2013). ROS, autophagy, mitochondria and cancer: Ras, the hidden master?. Mitochondrion, 13(3), 155-162.
- Pole, A., Dimri, M., Dimri, G. P. (2016). Oxidative stress, cellular senescence and ageing. AIMS Molecular Science, 3(3).
- Hekimi, S., Lapointe, J., and Wen, Y. (2011). Taking a good look at free radicals in the aging process. Trends Cell Biol. 21, 569576
- Ana P. Gomes, Nathan L. Price, Alvin J.Y. Ling, Javid J. Moslehi, Magdalene K. Montgomery, Luis Rajman, James P. White, Joo S. Teodoro, Christiane D. Wrann, Basil P. Hubbard, Evi M. Mercken, Carlos M. Palmeira, Rafael de Cabo, Anabela P. Rolo, Nigel Turner, Eric L. Bell, David A. Sinclair (2013). Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell. Dec 19;155(7):1624-38.
- Ryan, M.T., Hoogenraad, N.J. (2007). Mitochondrial-Nuclear Communications. Annu. Rev. Biochem. 76:70122
- Lim, J.H., Lee, Y.M., Chun, Y.S., Chen, J., Kim, J.E., and Park, J.W. (2010). Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxiainducible factor 1alpha. Cell 38, 864878.
- Finley, L. W., Carracedo, A., Lee, J., Souza, A., Egia, A., Zhang, J., …, Pandolfi, P. P. (2011). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α Cancer cell, 19(3), 416-428.
- Gerhart-Hines, Z., Rodgers, J.T., Bare, O., Lerin, C., Kim, S.H., Mostoslavsky, R., Alt, F.W., Wu, Z., and Puigserver, P. (2007). Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 26, 19131923.
- Rodgers, J.T., Lerin, C., Haas, W., Gygi, S.P., Spiegelman, B.M., and Puigserver, P. (2005). Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113118.
- Fang, E.F., Scheibye-Knudsen, M., Brace, L.E., Kassahun, H., Tanima SenGupta, Nilsen, H., Mitchell, J.R., Croteau, D.L., Bohr, V.A. (2014). Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 157, 882896, May 8.
- Sahin, E., and DePinho, R.A. (2012). Axis of ageing: telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 13, 397404.
- Green, D.R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagyinflammation-cell death axis in organismal aging. Science 333, 1109 1112.
- Fontana, L., Partridge, L., and Longo, V.D. (2010). Extending healthy life spanfrom yeast to humans. Science 328, 321326
- Kenyon, C.J. (2010). The genetics of ageing. Nature 464, 504512.
- Slack, C., Giannakou, M.E., Foley, A., Goss, M., and Partridge, L. (2011). dFOXOindependent effects of reduced insulin-like signaling in Drosophila. Aging Cell 10 , 735748.
- Salih, D.A.M, Brunet, A. (2008). FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. Apr; 20(2): 126136.
- Price, N.L., Gomes, A.P., Ling, A.J., Duarte, F.V., Martin-Montalvo, A., North, B.J., Agarwal, B., Ye, L., Ramadori, G., Teodoro, J.S., et al. (2012). SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15, 675690.
- Alers, S., Lo¨ffler, A.S., Wesselborg, S., and Stork, B. (2012). Role of AMPKmTORUlk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 211.
- Anisimov, V.N., Berstein, L.M., Popovich, I.G., Zabezhinski, M.A., Egormin, P.A., Piskunova, T.S., Semenchenko, A.V., Tyndyk, M.L., Yurova, M.N., Kovalenko, I.G., and Poroshina, T.E. (2011). If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (Albany NY) 3, 148157.
- Mair, W., Morantte, I., Rodrigues, A.P., Manning, G., Montminy, M., Shaw, R.J., and Dillin, A. (2011). Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 470, 404408.
- Onken, B., and Driscoll, M. (2010). Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 5, e8758.
- Harrison, D.E., Strong, R., Sharp, Z.D., Nelson, J.F., Astle, C.M., Flurkey, K., Nadon, N.L., Wilkinson, J.E., Frenkel, K., Carter, C.S., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392395.
- Bai, P., Cant´o, C., Oudart, H., Bruny´anszki, A., Cen, Y., Thomas, C., Yamamoto, H., Huber, A., Kiss, B., Houtkooper, R.H., et al. (2011). Cell Metab. 13, 461468.
- McDonald, R. B., Ruhe, R. C. (2011). Aging and longevity: why knowing the difference is important to nutrition research. Nutrients, 3(3), 274-282.
- Tchkonia, T., Zhu, Y., Van Deursen, J., Campisi, J., Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. The Journal of clinical investigation, 123(3), 966-972.
- Wang, C., Jurk, D., Maddick, M., Nelson, G., Martin-Ruiz, C., and von Zglinicki, T. (2009). DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8, 311323
- Rossi, D.J., Bryder, D., Seita, J., Nussenzweig, A., Hoeijmakers, J., and Weissman, I.L. (2007). Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725729.
- Janzen, V., Forkert, R., Fleming, H.E., Saito, Y., Waring, M.T., Dombkowski, D.M., Cheng, T., DePinho, R.A., Sharpless, N.E., and Scadden, D.T. (2006). Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443, 421426.
- Ferron, S. R., Marques-Torrejon, M., Mira, H., Flores, I., Taylor, K., Blasco, M. A. and Farinas, I. (2009). Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci. 29, 14394-14407.
- Flores, I., Canela, A., Vera, E., Tejera, A., Cotsarelis, G. and Blasco, M. A. (2008). The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 22, 654-667.
- Beerman, I., Bock, C., Garrison, B. S., Smith, Z. D., Gu, H., Meissner, A. and Rossi, J. (2013). Proliferation-dependent alterations of the DNA methylation
- Ru be, C. E., Fricke, A., Widmann, T. A., Fu rst, T., Madry, H., Pfreundschuh, M. and Ru be, C. (2011). Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS ONE 6, e17487.
- Corces-Zimmerman, M. R., Hong, W.-J., Weissman, I. L., Medeiros, B. C. and Majeti, R. (2014). Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 111, 2548-2553
- Genovese, G., K¨ahler, A. K., Handsaker, R. E., Lindberg, J., Rose, S. A., Bakhoum, S. F., Chambert, K., Mick, E., Neale, B. M., Fromer, M. et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 24772487.
- Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., Lindsley, C. R., Mermel, C. H., Burtt, N., Chavez, A. et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488-2498.
- Jan, M., Snyder, T. M., Corces-Zimmerman, M. R., Vyas, P., Weissman, I. L., Quake, S. R. and Majeti, R. (2012). Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 4, 149 ra 118.
- Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin, J., Liu, Y., Zhang, D., Scadden, D.T., and Chen, D. (2013). SIRT3 reverses aging-associated degeneration. Cell Rep. 3, 319327.
- Schultz, M.B., Sinclair, D.A. (2016) When stem cells grow old : phenotypes and mechanism of stem cell aging. Development 143, 3-14.
- Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010 Oct 14;6(10):e1001159.
- Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T Jr, Jones DL, Walker DW. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 2011 Nov 2;14(5):623-34.
- Villeda, S. A., Plambeck, K. E., Middeldorp, J., Castellano, J. M., Mosher, K. I., Luo, J., … Wabl, R. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nature medicine, 20(6), 659-663.
- Lavasani, M., Robinson, A.R., Lu, A., Song, M., Feduska, J.M., Ahani, B., Tilstra, J.S., Feldman, C.H., Robbins, P.D., Niedernhofer, L.J., and Huard, J. (2012). Musclederived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nat. Commun. 3, 608
- Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 7991.
- Toma s-Loba, A., Flores, I., Ferna ndez-Marcos, P.J., Cayuela, M.L., Maraver, A., Tejera, A., Borra s, C., Matheu, A., Klatt, P., Flores, J.M., et al. (2008). Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 135, 609622.
- Salminen, A., Kaarniranta, K., Kauppinen, A. (2012). Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 4, 166175
- Morley, J.E., Baumgartner, R.N. (2004). Cytokine-Related Aging Process Journal of Gerontology: MEDICAL SCIENCES, Vol. 59A, No. 9, 924929
- Ferrucci, L., Harris, T.B., Guralnik, J.M., Tracy, R.P., Corti, M.C., Cohen, H.J., Penninx, B., Pahor, M., Wallace, R., Havlik, R.J., 1999. Serum IL-6 level and the development of disability in older persons. J. Am. Geriatr. Soc. 47, 639646.
- Osorio, F.G., B´arcena, C., Soria-Valles, C., Ramsay, A.J., de Carlos, F., Cobo, J., Fueyo, A., Freije, J.M., and L´opez-Ot´ın, C. (2012). Nuclear lamina defects cause ATMdependent NF-kB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 26, 23112324.
- Tilstra, J.S., Robinson, A.R., Wang, J., Gregg, S.Q., Clauson, C.L., Reay, D.P., Nasto, L.A., St Croix, C.M., Usas, A., Vo, N., et al. (2012). NF-kB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122, 26012612.
- Adler, A.S., Sinha, S., Kawahara, T.L., Zhang, J.Y., Segal, E., and Chang, H.Y. (2007). Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 21, 32443257.
- Xie, J., Zhang, X., and Zhang, L. (2013). Negative regulation of inflammation by SIRT1. Pharmacol. Res. 67, 6067.
- Gillum, M.P., Kotas, M.E., Erion, D.M., Kursawe, R., Chatterjee, P., Nead, K.T., Muise, E.S., Hsiao, J.J., Frederick, D.W., Yonemitsu, S., et al. (2011). SirT1 regulates adipose tissue inflammation. Diabetes 60, 32353245.
- Yao, H., Chung, S., Hwang, J.W., Rajendrasozhan, S., Sundar, I.K., Dean, D.A., McBurney, M.W., Guarente, L., Gu, W., Ro¨nty, M., et al. (2012). SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Invest. 122, 20322045.
- Zhang, Z., Lowry, S.F., Guarente, L., and Haimovich, B. (2010). Roles of SIRT1 in the acute and restorative phases following induction of inflammation. J. Biol. Chem. 285 , 4139141401.
- Rothgiesser, K.M., Erener, S., Waibel, S., Lu¨scher, B., and Hottiger, M.O. (2010). SIRT2 regulates NF-kB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 123, 42514258.
- Pont, A.R., Sadri, N., Hsiao, S.J., Smith, S., and Schneider, R.J. (2012). mRNA decay factor AUF1 maintains normal aging, telomere maintenance, and suppression of senescence by activation of telomerase transcription. Mol. Cell 47, 515.
- Doles, J., Storer, M., Cozzuto, L., et al., (2012). Age-associated inflammation inhibits epidermal stem cell function. Genes Dev.
- Liu, N., Landreh, M., Cao, K., Abe, M., Hendriks, G.J., Kennerdell, J.R., Zhu, Y., Wang, L.S., and Bonini, N.M. (2012). The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482, 519523.
- Shen, Y., Wollam, J., Magner, D., Karalay, O., and Antebi, A. (2012). A steroid receptor-microRNA switch regulates life span in response to signals from the gonad. Science 338, 14721476.
- Smith-Vikos, T., and Slack, F.J. (2012). MicroRNAs and their roles in aging. J. Cell Sci. 125, 717.
- Boulias, K., and Horvitz, H.R. (2012). The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF- 16/FOXO. Cell Metab. 15, 439450.
- Toledano, H., DAlterio, C., Czech, B., Levine, E., and Jones, D.L. (2012). The let-7Imp axis regulates ageing of the Drosophila testis stem-cell niche. Nature 485, 605610.
- Ugalde, A.P., Espan˜ol, Y., and Lo´pez-Ot´ın, C. (2011). Micromanaging aging with miRNAs: new messages from the nuclear envelope. Nucleus 2, 549555.
- Benetti, R., Gonzalo, S., Jaco, I., Muoz, P., Gonzalez, S., Schoeftner, S., … and Li, E. (2008). A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nature structural and molecular biology, 15(3), 268-279.
- Mitomo, S., Maesawa, C., Ogasawara, S., Iwaya, T., Shibazaki, M., et al., 2008. Downregulation of miR-138 is associated with overexpression of human telomerase reverse transcriptase protein in human anaplastic thyroid carcinoma cell lines. Cancer Sci. 99 , 280286.
- Chen, Y.F., Kao, C.H., Chen, Y.T., Wang, C.H., Wu, C.Y., et al., (2009). Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 23, 11831194.
- Lee J., Kemper, J.K.(2010) Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging, 2-8.
- Card D.A., Hebbar P.B., Li L., Trotter K.W., Komatsu Y., et al. (2008). Oct4/Sox2regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 28, 64266438.
- Hackl, M., Brunner, S., Fortschegger, K., Schreiner, C., Micutkova, L., et al. (2010). miR- 17, miR-19b, miR-20a and miR-106a are down-regulated in human aging. Aging Cell 9, 291296
- Li, G., Luna, C., Qiu, J., Epstein, D.L., Gonzalez, P. (2009). Alterations in miRNA expression in stress-induced cellular senescence. Mech. Ageing Dev. 130, 731741.
- Fontana, L., Fiori, M.E., Albini, S., Cifaldi, L., Giovinazzi, S., et al. (2008). Antagomir17- 5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One 3, e2236.
- Taganov, K.D., Boldin, M.P., Chang, K.J., Baltimore, D., 2006. NF-κB-dependent induction of miRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103, 12481 12486.
- Iliopoulos, D., Hirsch, H.A., Struhl, K. (2009). An epigenetic switch involving NFκB, Lin28, Let-7 MiRNA, and IL6 links inflammation to cell transformation. Cell 139 , 693706
- Quinn, S. R., O’Neill, L. A. (2011). A trio of microRNAs that control Toll-like receptor signalling. International immunology, 23(7), 421-425.
- Olivieri, F., Rippo, M. R., Procopio, A. D., Fazioli, F. (2014). Circulating inflammamiRs in aging and age-related diseases. The origin, function and diagnostic potential of extracellular microRNA in human body fluids, 106.
- Boldin, M. P., Baltimore, D. (2012). MicroRNAs, new effectors and regulators of NFB. Immunological reviews, 246(1), 205-220.
- Steinmann, G. G., Klaus, B., Mu¨llerHermelink, H. K. (1985). The involution of the ageing human thymic epithelium is independent of puberty. Scandinavian journal of immunology, 22(5), 563-575.
- Steinmann, G. G. (1986). Changes in the human thymus during aging. In The Human Thymus (pp. 43-88). Springer Berlin Heidelberg.
- Lynch, H. E., Goldberg, G. L., Chidgey, A., Van den Brink, M. R., Boyd, R., Sempowski, G. D. (2009). Thymic involution and immune reconstitution. Trends in immunology, 30(7), 366-373.
- Weksler, M. E., Pawelec, G., Franceschi, C. (2009). Immune therapy for age-related diseases. Trends in immunology, 30(7), 344-350.
- Chen, W. H., Kozlovsky, B. F., Effros, R. B., Grubeck-Loebenstein, B., Edelman, R., Sztein, M. B. (2009). Vaccination in the elderly: an immunological perspective. Trends in immunology, 30(7), 351-359.
- Dowling, M. R., Hodgkin, P. D. (2009). Why does the thymus involute? A selectionbased hypothesis. Trends in immunology, 30(7), 295-300.
- Swaab, D. F., Hofman, M. A., Lucassen, P. J., Purba, J. S., Raadsheer, F. C., Van de Nes, J. A. P. (1993). Functional neuroanatomy and neuropathology of the human hypothalamus. Anatomy and embryology, 187(4), 317-330.
- Bartke, A. (2011). Pleiotropic effects of growth hormone signaling in aging. Trends Endocrinol. Metab. 22, 437442.
- Zhang, G., Li, J., Purkayastha, S., Tang, Y., Zhang, H., Yin, Y., Li, B., Liu, G., and Cai, D. (2013). Hypothalamic programming of systemic ageing involving IKK-b, NF-kB and GnRH. Nature 497, 211216.
- Satoh, A., Brace, C. S., Rensing, N., Cliften, P., Wozniak, D. F., Herzog, E. D., … Imai, S. I. (2013). Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell metabolism, 18(3), 416-430.
- Imai, S. I., Guarente, L. (2014). NAD+ and sirtuins in aging and disease. Trends in cell biology, 24(8), 464-471.
- Chang, H., Guarente, L. (2013). SIRT1 and other sirtuins in metabolism Trends in Endocrinology and Metabolism, 18.
- Kumar, S., Lombard, D. B. (2015). Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxidants and redox signaling, 22(12), 1060-1077.
- Rodier, F., Campisi, J., Bhaumik, D. (2007). Two faces of p53: aging and tumor suppression. Nucleic acids research, 35(22), 7475-7484.
- Maiuri, M.C., Galluzzi L., Morselli E., Kepp O., Malik S.A., Kroemer G. (2010). Autophagy regulation by p53 Current Opinion in Cell Biology, 22:181185
- Kroemer, G., Marin˜o, G., Levine, B. (2010). Autophagy and the Integrated Stress Response. Molecular Cell 40.
- Matoba, S., Kang, J.G., Patino, W.D., Wragg, A., Boehm, M., Gavrilova, O., Hurley, P.J., Bunz, F., Hwang, P.M. (2006). p53 Regulates Mitochondrial Respiration. Science 312, 1650.
- Ferbeyre, G., Lowe, S. W. (2002). Ageing: The price of tumour suppression?. Nature, 415(6867), 26-27.
- Morimoto, R.I., Cuervo, A.M. (2014). Proteostasis and the aging proteome in health and disease. J. Gerontol. A Biol. Sci. Med. Sci. 69 (suppl. 1), S33S38
- Verdin, E., Hirschey, M. D., Finley, L. W., Haigis, M. C. (2010). Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends in biochemical sciences, 35(12), 669-675.
- Palikaras, K., Tavernarakis, N. (2012). Mitophagy in neurodegeneration and aging. Frontiers in genetics, 3, 297.
- Canto, C., Auwerx, J. (2009). PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current opinion in lipidology, 20(2), 98.
- Shih, J., Donmez, G. (2013). Mitochondrial sirtuins as therapeutic targets for agerelated disorders. Genes and cancer, 4(3-4), 91-96.
- Fernandez-Marcos, P.J., and Auwerx, J. (2011). Regulation of PGC-1a, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884S890 S.
- Iyer, N. V., Kotch, L. E., Agani, F., Leung, S. W., Laughner, E., Wenger, R. H., … Semenza, G. L. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1. Genes and development, 12(2), 149-162.
- Semenza, G. L. (2003). Targeting HIF-1 for cancer therapy. Nature reviews cancer, 3(10), 721-732.
- Haigis, M. C., Guarente, L. P. (2006). Mammalian sirtuinsemerging roles in physiology, aging, and calorie restriction. Genes and development, 20(21), 2913-2921.
- Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., … Sinclair, D. A. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science, 305(5682), 390-392.










