

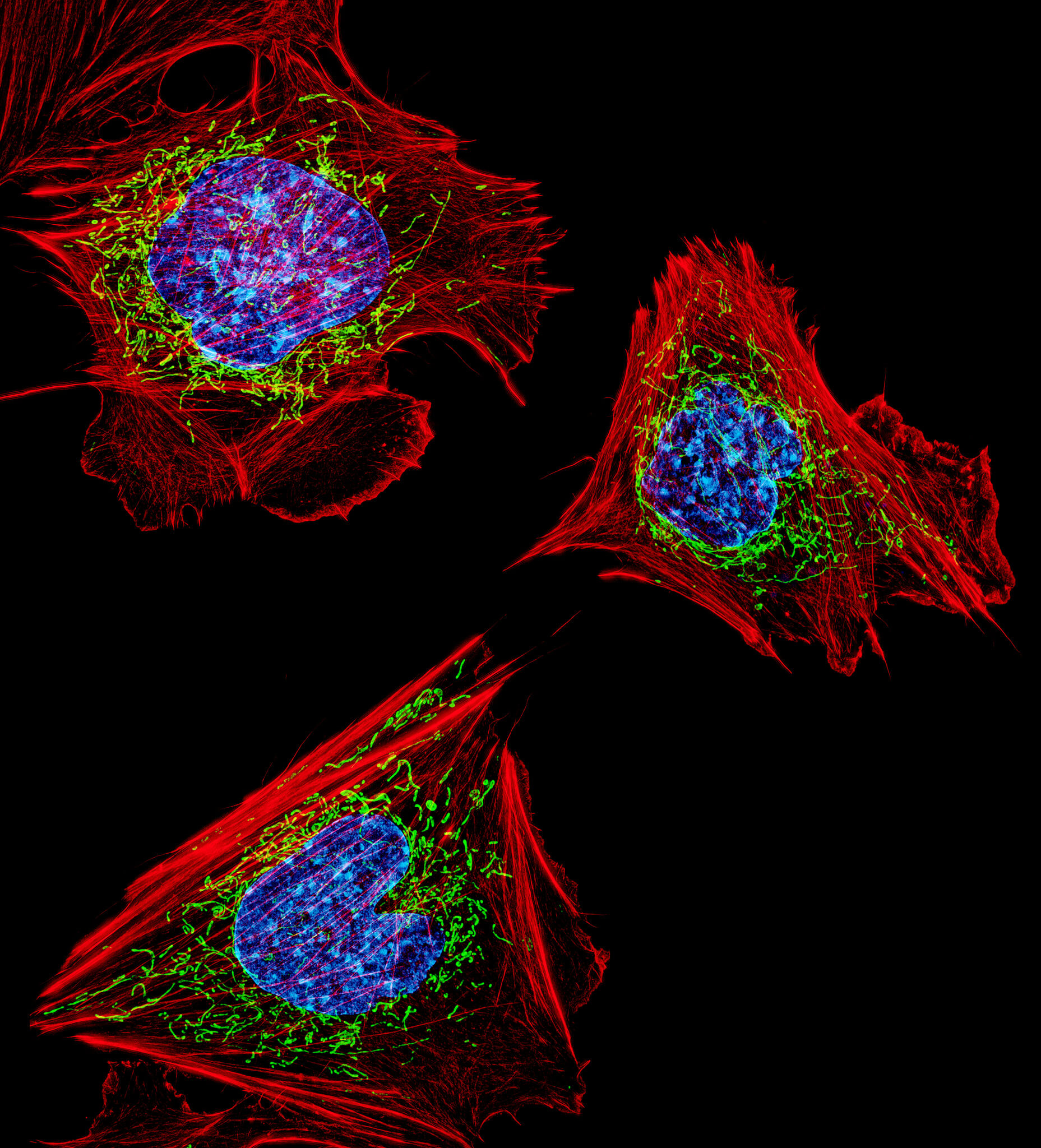
Comment stresser une cellule
Comme nous l’avons vu dans la partie précédente, il existe plusieurs types de stress et la cellule dispose de réponses variées pour y faire face. Les réponses les plus classiques font intervenir des organites distincts dont la mitochondrie, qui joue un rôle central dans la régulation de l’apoptose, le réticulum, qui permet la médiation de l’UPR (Unfolded Protein Response), le noyau, dans lequel les lésions de l’ADN peuvent enclencher un stress, ou encore les peroxysomes qui ont un rôle important dans la réponse au stress oxydant.
Jetons un oeil sur tous ces mécanismes pour mieux comprendre comment le stress cellulaire peut impacter nos cellules !
La mitochondrie : la première cible du stress cellulaire
La mitochondrie est un organite un peu curieux. Sa présence dans le cytoplasme de nos cellules est le résultat d’une fusion d’une vieille bactérie avec nos cellules (théorie endosymbiotique) il y a plus de 2 milliards d’années [1]. Elle possède son propre ADN (noté ADNmt pour ADN mitochondrial) et se transmet d’une cellule mère à une cellule fille grâce à une augmentation de sa taille suivie d’une fission. Elle a un rôle central dans notre métabolisme énergétique puisqu’elle gère la production d’ATP et de NADH notamment, deux cofacteurs indispensables au fonctionnement de nos enzymes. Sa transmission se fait uniquement par la mère, grâce aux mitochondries présentes dans les ovules, les mitochondries des spermatozoïdes ne passant pas la membrane de l’ovocyte lors de la fécondation [1]. Il existe des maladies mitochondriales pouvant mener à des retards mentaux, des dysfonctions métaboliques ou des problèmes cardiaques, toujours d’origine maternelle.
Dans un contexte physiologique, la mitochondrie assure différents rôles et est la cible privilégiée (et la plus étudiée) du stress cellulaire.
Phosphorylation oxydative et coenzymes
Lorsqu’une cellule fonctionne normalement, elle utilise des coenzymes (ou cofacteurs), de petites molécules permettant l’action des enzymes. Parmi ces coenzymes, on retrouve l’ATP et le NADH. Lorsque les enzymes travaillent, elles utilisent ces cofacteurs afin d’apporter assez d’énergie à la réaction qu’elles rendent possible et permettent son accélération. Lors de ce phénomène, l’ATP est transformé en ADP (il perd un groupe phosphate) et afin de le recycler, la mitochondrie va mettre en œuvre tout un système. Sans rentrer dans le détail de processus métaboliques complexes, la mitochondrie va rajouter un groupement phosphate à l’ADP pour le transformer en ATP et permettre sa réutilisation. Cette action est rendue possible par des mécanismes d’oxydoréduction successifs qui utilisent l’énergie libérée par l’utilisation du NADH (transformé en NAD+) et à l’action de l’ATP-synthase.
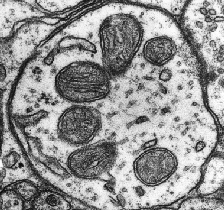
Bien qu’indispensable au fonctionnement cellulaire, la phosphorylation oxydative entraine la formation de radicaux libres et d’espèces réactives de l’oxygène (ROS) [2]. On observe trois formes majeures de ROS : le peroxide d’hydrogène (H2O2), le radical ·OH et l’ion superoxyde O2·− [3]. De ces trois espèces, la plus réactive est ·OH, qui peut modifier l’ADN nucléaire et mitochondrial et casser ses brins [3]. Pour combattre ce stress oxydant, nos cellules vont développer diverses stratégies impliquant des machineries antioxydantes (voir partie 1: les différents types de stress cellulaires) [4]. En temps normal, la mitochondrie et la cellule sont capable de maintenir un équilibre entre la formation des ces molécules et leur élimination. Cependant, lors d’un évènement extérieur ou d’une exposition à des médicaments connus pour augmenter la synthèse de ROS, cette balance peut pencher dans le mauvais sens et produire un stress cellulaire oxydant [5].
La régulation de l’apoptose
L’apoptose est un mécanisme de mort cellulaire programmée enclenché par la cellule lorsque les conditions garantissant sa survie ne sont plus réunies. Elle peut être activée par différents phénomènes, mais le régulateur central de l’apoptose demeure la mitochondrie. L’activation du signal apoptotique suite à un stress cellulaire a lieu via la voie des caspases, et plus particulièrement les caspases 3, 8 et 9. La caspase-8 est la première à être activée, grâce à une voie de signalisation complexe, mais bien connue, faisant intervenir des kinases (une classe spécifique d’enzymes) dont Akt, ERK1/2, p38 et JNK [6] elles-mêmes activées par les protéines de la famille Bcl-2.
Une fois activée, la caspase-8 va transmettre le signal soit directement, via la caspase-3, soit indirectement en passant par la mitochondrie. La mitochondrie va en effet pouvoir réagir à la caspase-8, qui va indirectement enclencher la perméabilisation de sa membrane [7]. Le cytochrome c va pouvoir sortir de la mitochondrie, où il est normalement séquestré, et enclencher de nouvelles voies pro-apoptotiques. La synthèse de ROS est parallèlement augmentée, créant une boucle favorisant la mort cellulaire [8]. En plus de ces mécanismes, des molécules déclenchant un stress cellulaire peuvent provoquer directement la perméabilisation de la membrane mitochondriale, sans passer par l’activation de la caspase-8. La présence de petits trous (pores) dans la mitochondrie va perturber le potentiel de membrane et provoquer une perte de l’homéostasie cellulaire : la synthèse d’ATP n’aura plus lieu, le NADH va être oxydé et des ROS vont être synthétisés en excès. En réponse à ces phénomènes, la mitochondrie va relarguer plusieurs protéines, dont la plus connue est AIF (Apoptosis-Inducing Factor), qui vont aller se fixer au niveau de l’ADN pour le cliver et provoquer l’apoptose [9].
Le nombre de stimulus pouvant enclencher une réponse mitochondriale au stress cellulaire est impressionnant. Des années de recherche ont bien entendu participé à expliquer une partie de ces mécanismes, et nous savons aujourd’hui le rôle prépondérant des ROS et de la mitochondrie. Cependant, malgré l’évidence physiologique de l’implication du stress oxydant dans de multiples pathologies, comme l’Alzheimer, les cancers ou le diabète, le mode d’action des ROS reste encore incomplètement élucidé.
Le stress du réticulum
Le réticulum est un organite qui permet la synthèse, la modification et le repliement des protéines. On distingue deux types de réticulums: le rugueux, qui possède des ribosomes et traduit l’ARN en protéines, et le lisse, qui permet les modifications post-traductionnelles. C’est un élément indispensable à la bonne fonction de nos cellules et l’une des cibles favorites du stress cellulaire.
La dérégulation du calcium
La production de protéines qui a lieu dans le réticulum est un mécanisme oxydatif et nécessite un environnement spécifique permettant la formation de structures dans les protéines (pont disulfures, glycosylation…), indispensables pour leur repliement. Il est également très riche en calcium et en protéines chaperons [10]. Du fait de cet environnement oxydatif, le réticulum relargue des ROS qui, lorsque les protéines se replient mal ou que la cellule est soumise à un stress, sont produites en excès. Elles peuvent par la suite aller se fixer sur des canaux membranaires. Ces structures sont, comme leur nom l’indique, de petits tunnels dans lesquels vont pouvoir passer toutes sortes de molécules, à commencer par des ions, comme le calcium (Ca2+). Le réticulum va donc, suite à la régulation des ses canaux calciques par les ROS, relarguer du Ca2+ dans le cytoplasme, déclencher des signaux de stress et engendrer une réponse cellulaire [11]. Un de ces signaux consiste à envoyer le Ca2+ dans la mitochondrie qui, lorsque la concentration calcique augmente, va produire de nouveaux ROS et entretenir une boucle d’auto-activation du stress cellulaire.
En plus de cela, le trop-plein de calcium va entrainer l’activation de voies de synthèse amenant à la production de cytokines pro-inflammatoires (du type NFκB) et de protéines chaperons (afin de faire face à l’abondance de protéines mal repliées) : c’est ce qu’on appelle l »overload reticulum response » [12].
Le mauvais repliement des protéines
Pour compléter le cercle vicieux du stress cellulaire, lorsque le réticulum est stressé, produit beaucoup de ROS et relargue du calcium, activant ainsi des voies de signalisation pro-stress, il entretient également son propre stress en produisant de plus en plus de protéines mal repliées. Cela enclenche l’UPR (« Unfolded Protein Response »), un mécanisme développé dans la première partie de ce dossier, qui va obliger la cellule à choisir entre la voie de survie ou l’activation de l’apoptose.
Ainsi, le réticulum peut répondre de plusieurs manières à un stress cellulaire [13]
- une atténuation de la réponse transcriptionnelle passant par une régulation des gènes, afin de réduire le nombre de protéines produites,
- la régulation à la hausse de gènes codant pour les protéines chaperons, via la régulation calcique, afin de replier correctement le protéines qui ne le sont pas,
- la synthèse de cytokines pro-inflammatoires, via la régulation calcique,
- la mise en place de l’apoptose, lorsque les trois premières options ont échoué.
Stress du noyau et mécanismes génotoxiques
La régulation génique est indispensable à la réponse au stress cellulaire et passe par différents mécanismes, dont le stress du réticulum et de la mitochondrie. Elle permet, en effet, une adaptation des protéines produites et un ajustement des enzymes synthétisées, afin de répondre aux demandes spécifiques de la cellule au moment de l’agression. Il existe cependant un autre type de stress provenant du noyau lui-même. L’ADN est une cible comme une autre lorsque le stress est concerné, et nombreuses sont les substances chimiques ou les événements extérieurs pouvant causer un dommage sur sa structure. L’ADN est très sensible aux traitements chimiothérapeutiques, à l’irradiation ou à des éléments génotoxiques tels que les UV. Il est le plus fragile lorsqu’il est en cours de transcription ou de réplication, des étapes cellulaires normales au cours desquelles il se présente sous sa forme simple brin et où il peut être cassé. Fort heureusement, il existe toute une machinerie permettant sa réparation: la cassure est reconnue par des enzymes spécifiques (ERCC1-XPF et XPG) qui peuvent alors agir et réparer les petites lésions simple brin [14]. En revanche, lorsque la cassure a lieu sur le double brin de l’ADN, une toute autre réponse se met en place: des enzymes sont recrutées (notamment le complexe MRN) pour enclencher une voie de signalisation centrale, celle de p53 [15, 16]. p53 est une protéine importante dans la régulation de l’apoptose et son activation va déclencher la synthèse de facteurs pro-apoptotiques, mais aussi stimuler la mitochondrie et favoriser la mort cellulaire.
La prolifération des péroxysomes

Jamais entendu parler des peroxysomes ? C’est normal, on en parle peu, bien que leur rôle soit important pour nos cellules car ils participent à leur détoxification, via la dégradation de certains acides gras et alcools. Seul bémol, lorsqu’ils dégradent ces éléments, ils produisent de l’H2O2 . Dans une certaine mesure, les catalases présentes dans le peroxysome peuvent convertir l’H2O2 en H2O et O2, permettant ainsi un recyclage de cette molécule toxique, tout en réinjectant de l’oxygène dans le métabolisme cellulaire [17]. Sur le papier, c’est beau. Cependant, nous ne comprenons pas très bien comment les peroxysomes maintiennent un équilibre entre les ROS produits et celles dégradées. De plus, en cas de problème au niveau des peroxysomes, l’H2O2 est largement relarguée dans le cytoplasme augmentant significativement le stress cellulaire oxydant [17].
Les peroxysomes peuvent également se multiplier via l’action des activateurs de prolifération sur leur récepteurs nucléaires, les PPAR (Peroxysome Proliferation Activator Receptors). Les PPARs peuvent être activés par différents signaux environnementaux, nutritionnels ou encore inflammatoires : on dénote par exemple les prostaglandines, certains acides gras ou le leukotriène pro-inflammatoire, l’acide arachidonique, certains hypo-lipidémiants et AINS. Il existe trois formes de PPARs, α, β et ϒ, se ressemblant beaucoup, mais dont l’expression tissulaire peut varier. Lorsqu’ils sont activés, ils créent des hétérodimères avec un autre récepteur capable de se lier à l’ADN et régulent ainsi l’expression de certaines protéines, dont le gène contient un promoteur avec une séquence de reconnaissance de PPAR (peroxisome proliferator response element) [18]. Parmi ces gènes, on retrouve évidemment ceux impliqués dans la prolifération des peroxysomes, mais aussi certains gènes codant pour des protéines de la chaine respiratoire mitochondriale ou des anti-oxydants.
Tous ces organites touchés par le stress cellulaire en sont profondément modifiés et déterminent la survie de la cellule dans sa totalité. Lors d’un stress fort, il n’est pas rare d’observer la mort de milliers de cellules en parallèle pouvant avoir un impact sur notre santé. Lors du vieillissement, ces processus sont de plus en plus fréquents et expliquent partiellement pourquoi notre corps accuse son âge !
Quelques définitions
Caspases : les caspases sont des protéines jouant un rôle central dans la mort cellulaire programmée (apoptose majoritairement) et l’inflammation. Ce sont des enzymes capables d’activer beaucoup de voies de signalisation via l’activité cystéine protéase de leur site actif.
Potentiel de membrane : les membranes cellulaires sont des bicouches phospholipidiques, c’est à dire qu’elles sont formées de deux feuillets de lipides modifiés. Il y a donc une membrane interne, un espace inter-membranaire et une membrane externe. A l’intérieur et à l’extérieur de ces différentes membranes, des ions (positifs ou négatifs) sont présents. Afin d’éviter qu’ils ne rentrent et sortent comme ils le souhaitent et dérégulent l’homéostasie cellulaire (maintien du pH, de la fonction des enzymes, de la synthèse des protéines…), les membranes sont imperméables et gèrent l’entrée et la sortie de ces ions, créant ainsi un potentiel de membrane, c’est à dire la différence entre le potentiel à l’intérieur de la membrane interne et le potentiel à l’extérieur de la membrane externe.
AINS : Anti-Inflammatoire Non Stéroidien, tel que l’aspirine ou l’ibuprofène.
Tout notre dossier Stress cellulaire et vieillissement :
Stress cellulaire et vieillissement, binôme obligatoire ?

Bien que communément acquis par la communauté scientifique comme un facteur de risque et un déterminant de certaines maladies, beaucoup de questionnements subsistent vis à vis du stress cellulaire. Son rôle dans les pathologies liées au vieillissement et le vieillissement lui-même est toujours en cours d’étude.
Partie 1 : Les différents types de stress cellulaires
 Quand on parle de stress cellulaire, nous pouvons faire référence à beaucoup de mécanismes différents, des stress mécanique, toxique, chimique, thermique, osmotique, ionisant… tous participant au vieillissement de nos cellules.
Quand on parle de stress cellulaire, nous pouvons faire référence à beaucoup de mécanismes différents, des stress mécanique, toxique, chimique, thermique, osmotique, ionisant… tous participant au vieillissement de nos cellules.
Partie 2 : Les organites touchés par le stress cellulaire
 Le stress cellulaire est transmis par différents organites dans nos cellules, notamment la mitochondrie, le réticulum et le noyau, pour ne citer qu’eux. Leurs réponses varient et peuvent donner deux options à la cellule stressée: survivre et s’adapter ou mourir.
Le stress cellulaire est transmis par différents organites dans nos cellules, notamment la mitochondrie, le réticulum et le noyau, pour ne citer qu’eux. Leurs réponses varient et peuvent donner deux options à la cellule stressée: survivre et s’adapter ou mourir.
Partie 3 : Comment maîtriser le stress cellulaire
 Afin de ralentir le vieillissement de nos cellules, il est indispensable de maîtriser le stress cellulaire. Dans cette partie, nous ferons le tour des techniques et/ou médicaments actuellement plébiscités pour traiter le stress cellulaire sous toutes ses formes.
Afin de ralentir le vieillissement de nos cellules, il est indispensable de maîtriser le stress cellulaire. Dans cette partie, nous ferons le tour des techniques et/ou médicaments actuellement plébiscités pour traiter le stress cellulaire sous toutes ses formes.
Références
[1] Friedman JR & Nunnari J, Mitochondrial form and function, Nature, 2014;505:335–343
[2] Turrens JF, Boveris A, Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 1980;191:421–427
[3] Matés JA, Segura FJ, Alonso JM, Javier M, Oxidative stress in apoptosis and cancer: an update. Arch Toxicol. 2012;86:11, pp 1649–1665
[4] Masutani H, Yodoi J (2002) Thioredoxin. Overview. Methods Enzymol 347:279–286
[5] Farrugia G, Balzan R, Oxidative stress and programmed cell death in yeast. Front Oncol 2012;2:64
[6] Gabai VL, Yaglom JA, Volloch V et al., Hsp72-mediated suppression of c-Jun N-terminal kinase is implicated in development of tolerance to caspase-independent cell death. Mol Cell Biol 2000;20:6826–6836
[7] Barnhart BC, Alappat EC, Peter ME, The CD95 type I/type II model. Semin Immunol 2003;15:185–193
[8] Circu ML, Aw TY, Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 2010;48:749–762
[9] Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
[10] Schröder M, Kaufman RJ, ER stress and the unfolded protein response. Mutat Res. 2005 Jan 6; 569(1-2):29-63
[11] Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. Journal of Clinical Investigation. 2005;115(10):2656-2664
[12] Kong L, Li S, Huang M, et al. The Roles of Endoplasmic Reticulum Overload Response Induced by HCV and NS4B Protein in Human Hepatocyte Viability and Virus Replication. Shoukry NH, ed. PLoS ONE. 2015;10(4):e0123190
[13] Oyadomari S, Mori M, Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death and differentiation. 2004, 11. 381-9
[14] L. Staresincic, A. F. Fagbemi, J. H. Enzlin et al., Coordination of dual incision and repair synthesis in human nucleotide excision repair, EMBO Journal, 2009, vol. 28, no. 8, pp. 1111–1120
[15] M. Christmann, M. T. Tomicic, W. P. Roos, and B. Kaina, Mechanisms of human DNA repair: an update, Toxicology, 2003, vol. 193, no. 1-2, pp. 3–34
[16] J. W. Harper and S. J. Elledge, The DNA damage response: ten years after, Molecular Cell, 2007 vol. 28, no. 5, pp. 739–745
[17] Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J, Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem 2004;266:37–56
Dr. Marion Tible
Author/Reviewer
Auteure/Relectrice
Marion Tible has a PhD in cellular biology and physiopathology. Formerly a researcher in thematics varying from cardiology to neurodegenerative diseases, she is now part of Long Long Life team and is involved in scientific writing and anti-aging research.
More about the Long Long Life team
Marion Tible est docteur en biologie cellulaire et physiopathologie. Ancienne chercheuse dans des thématiques oscillant de la cardiologie aux maladies neurodégénératives, elle est aujourd’hui impliquée au sein de Long Long Life pour la rédaction scientifique et la recherche contre le vieillissement.
En savoir plus sur l’équipe de Long Long Life
Dr Guilhem Velvé Casquillas

Author/Reviewer
Auteur/Relecteur
Physics PhD, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
More about the Long Long Life team
Docteur en physique, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
En savoir plus sur l’équipe de Long Long Life










