


Longévité : une étude
Les causes biologiques du vieillissement et de la limitation de l’espérance de vie : plus on en apprend sur le vieillissement, moins il nous semble inéluctable. Il est frappant de constater que la vitesse de vieillissement varie considérablement d’une espèce à une autre, d’un individu à un autre, au sein d’une même espèce. Certaines espèces vieillissent si lentement que leurs représentants meurent systématiquement de famine, de maladie, d’accident ou de prédation, avant même d’avoir montré tout signe de sénescence. De ce fait, il est tentant d’imaginer qu’une meilleure connaissance des éléments qui déterminent la durée de vie pourrait nous permettre de ralentir le vieillissement.

Pourrait-on inverser le vieillissement ?
Parmi les scientifiques, différentes perceptions du vieillissement font rage. Est-ce que le vieillissement suit une structure spécifique provoquée par les gènes ? Se produit-il par hasard, par coïncidence ? Selon la théorie de l’évolution, tous les processus biologiques sont génétiquement programmés du début de la vie au stade de la maturité reproductive. Pour le reste, les biologistes sont en désaccord quant à savoir si les gènes provoquent un état de vieillissement avancé de l’organisme ou si le vieillissement des cellules souches à partir d’une accumulation aléatoire de lésions affecte la fidélité moléculaire [1]. Ainsi, le vieillissement défie toute définition acceptée universellement.
Dr L. Hayflick
Il déplore le fait que cela conduise à des échecs dans la communication et l’interprétation des résultats de recherche [2]. Il préconise une approche stochastique du vieillissement et une vision déterministe de la durée de vie [2] [3]:

« L’ENTROPIE EXPLIQUE LE VIEILLISSEMENT, LE DÉTERMINISME GÉNÉTIQUE EXPLIQUE LA DURÉE DE VIE, ET UNE TERMINOLOGIE INDÉTERMINÉE EXPLIQUE UNE CONFUSION ENTRE LES DEUX. »
L. Hayflick
Grâce à la théorie de la maintenance, on a remarqué que le vieillissement se manifestait lorsque le système de réparation cellulaire n’était plus en mesure de préserver l’équilibre qui existait avant la maturité reproductive. Certains affirment que cet équilibre est finalement voué à l’échec en raison d’une entropie croissante [8].
Les partisans du soma jetable et de la théorie de la pléiotropie antagoniste voient le vieillissement comme un effet secondaire négatif mais inévitable des fonctions nécessaires. Williams, qui est à l’origine de cette théorie, a écrit que la sélection naturelle pouvait favoriser la forme physique parmi les jeunes, au détriment de la forme à l’âge adulte [9].
« ON PEUT AFFIRMER QUE LA SÉLECTION NATURELLE FAVORISE PLUS LES JEUNES QUE LES PERSONNES ÂGÉES DÈS LORS QU’UN CONFLIT D’INTÉRÊT APPARAÎT. »
G. Williams

D’autres théories suggèrent également que le vieillissement serait le résultat d’un compromis entre une certaine qualité procurant un avantage chez les jeunes et un désavantage chez les personnes âgées [10]. Mais pour quelle raison l’expression d’un gène change-t-elle considérablement lors du vieillissement? Inspirés par la théorie controversée de la sélection de groupe, des scientifiques ont montré que le vieillissement pourrait favoriser la survie d’un groupe d’individus tout en mettant en péril la forme physique individuelle. Cet argument soutient les théories du vieillissement programmé. Comme Wilson l’avait remarqué en 1974 [11]:
« [PARLER DE PROGRAMMATION] C’EST FAIRE L’HYPOTHÈSE QU’IL Y A DES AVANTAGES DIRECTS OU INDIRECTS À LIMITER LA DURÉE DE VIE par le VIEILLISSEMENT. »
Wilson
Pour en savoir plus sur ce débat et ses implications :
Le vieillissement peut être traité à différents niveaux. Tout d’abord, il y a une perte de la fidélité moléculaire – certains parlent de vieillissement moléculaire. Ensuite survient un vieillissement individuel des cellules, connu sous le nom de sénescence. La sénescence des cellules est un mécanisme clé pour le développement, mais devient néfaste lorsqu’elle affecte la fonction des cellules souches et immunitaires, rendant ainsi l’homéostasie tissulaire difficile. Le vieillissement se caractérise donc par un changement de phénotype au niveau des tissus et des organes. L’hypothèse que le vieillissement pourrait être provoqué par des facteurs systémiques a récemment déclenché d’intenses recherches dans le monde scientifique.
Cet article examine les principales causes biologiques du vieillissement ciblées par thérapies expérimentales contre le vieillissement. Pour plus d’informations, veuillez lire The Hallmarks of Aging [12].

Les lésions du génome, une limite à la longévité ?
Les lésions au niveau de l’ADN s’accumulent tout au long de la vie, car de nombreux facteurs menacent la stabilité du génome. Ils comprennent des causes extrinsèques physiques, chimiques, des agents biologiques, ainsi que des menaces intrinsèques (ADN, erreurs de réplication, réactions hydrolytiques et dérivés réactifs de l’oxygène) [13]. Des défaillances dans les mécanismes de réparation de l’ADN accélèrent le vieillissement chez les souris et les humains, suggérant ainsi des liens de causalité entre une accumulation des lésions au niveau de l’ADN nucléaire et le vieillissement [13] [14] [15].

Lésions de l’ADN mitochondrial. L’implication des mutations de l’ADN mitochondrial (ADNmt) dans le processus de vieillissement a été prouvée : une détérioration de l’ADNmt via des mutations [16] ou une défection de l’ADN polymérase [17] [18] [19] peuvent engendrer un vieillissement prématuré, ou réduire la durée de vie.
L’ADNmT est-il plus ou moins vulnérable aux lésions que l’ADN nucléaire ? La controverse demeure. Par ailleurs, les nombreuses copies d’ADNmt dans chaque cellule atténueraient probablement les conséquences d’une lésion de l’ADNmt. Curieusement, le nombre de mitochondries diminue avec l’âge dans les cellules hépatiques des souris [20], des rats [21], et des humains [22] [23], ce qui correspond à une diminution du nombre de copies de l’ADNmt. Ceci expliquerait pourquoi l’accumulation des mutations au cœur du génome mitochondrial pourrait à terme devenir significatif dans le processus de vieillissement. Autre explication : l’accumulation des cellules contenant des niveaux élevés d’ADNmt mutant pourrait être un résultat inévitable des mécanismes normaux qui servent à maintenir les concentrations cellulaires d’ADNmt [25].
Lésions de l’ADN nucléaire. Les mutations somatiques et les anomalies chromosomiques sont plus susceptibles d’être observées chez les organismes vieux que chez les organismes jeunes. [26] [27] [28]. Les altérations de l’ADN peuvent à terme endommager des gènes essentiels ou des voies de transcription. Par conséquent, le dysfonctionnement cellulaire pourrait compromettre l’homéostasie tissulaire ou organismique, s’il n’est pas éradiqué par l’apoptose ou la sénescence. Les dysfonctionnements liés à l’âge au niveau de la lamina nucléaire pourraient également intensifier les lésions génomiques. [29]
De récentes études suggèrent que les méthodes de réparation de l’ADN nucléaire pourraient jouer un rôle dans le processus de vieillissement. Les protéines poly(ADP-ribose) polymérase (PARP) NAD+-dépendantes (1) sont souvent associées au mécanisme de réparation de l’ADN. D’autres études les ont également associées aux fonctions des mitochondries(4) et au métabolisme oxydant [30] [31]. L’activation d’une protéine PARP influence le métabolisme cellulaire au moyen d’altérations du métabolisme NAD+(1), d’activités liés à la PARylation (une altération post-translationnelle du PARP-dépendant) et la reprogrammation transcriptionnelle des cellules [31]. De façon générale, les inhibiteurs de PARP renforcent le métabolisme oxydatif et augmentent la quantité de mitochondries(4). Il faut noter qu’une amélioration de l’activité PARP a été observée chez les organismes âgés[32]. La PARP-1 est une enzyme nucléaire abondante activée par des lésions de l’ADN. Elle est impliquée dans des mécanismes de réparation. Son activation pourrait rapidement conduire à un appauvrissement du pool de NAD+ cytosolique(1), et provoquerait même une apoptose des cellules [33] en cas de lésions considérables au niveau de l’ADN.
Un autre processus de réponse aux lésions de l’ADN est réalisé par le suppresseur de tumeur p53(3). Il faut noter que la protéine p53 peut augmenter longévité en prévenant l’apparition d’un cancer, mais une activation accrue de p53 peut avoir des effets nuisibles. Elle favoriserait des aspects du processus de vieillissement en remettant en question l’homéostasie des tissus avec une sénescence excessive ou un signalement apoptotique (voir « Sénescence cellulaire » et « Rétrécissement des télomères »). [34]
Tant que les lésions au niveau de l’ADN s’accumulent avec l’âge chronologique, la consommation de NAD+(1) par les protéines PARP augmente. L’activité des sirtuines (2) est susceptible d’être perturbée par l’appauvrissement du pool de NAD+. En ce qui concerne la protéine p53 (3), sa surexpression chez les cellules vieillissantes pourrait être provoquée par l’activation de protéines PARP [35].
On peut supposer que l’accumulation de lésions au niveau de l’ADN pourrait jouer le rôle d’une horloge biologique activée par une entropie grandissante. Les lésions de l’ADN pourraient croître exponentiellement, car elles affectent à terme la reproduction du code génétique et le mécanisme de réparation, qui pourrait devenir donc moins efficace dans la reproduction fidèle de l’ADN. Avec une activation excessive de la protéine PARP, le mécanisme induit par celle-ci est destiné à protéger l’organisme contre les cellules endommagées de l’ADN et pourrait à terme appauvrir le pool (1) NAD+ et compromettre la fonction des sirtuines NAD+ dépendantes (2) [36]. Même si le rôle des sirtuines dans le processus de vieillissement n’est pas parfaitement connu, cette altération pourrait avoir de multiples conséquences sur le métabolisme des cellules.
Rétrécissement des télomères : la sénescence cellulaire programmée ?
La détérioration de l’ADN affecte le génome de façon presque aléatoire. Cependant, certaines zones chromosomiques sont particulièrement sensibles aux lésions associées au vieillissement [37]. Les ADN polymérases réplicatives ne reproduisent pas soigneusement la fin de structure moléculaire de l’ADN. Cette fonction est exclusivement effectuée par l’ADN polymérase spéciale connue sous le nom de télomérase. Chez l’être humain, les télomérases sont uniquement exprimées dans les cellules souches embryonnaires (leur permettant de se diviser à plusieurs reprises), dans certaines cellules souches adultes, et dans les cellules tumorales. La majeure partie des cellules de mammifères n’expriment pas de télomérase. Cela conduit à un manque progressif et cumulatif des extrémités des chromosomes pendant les cycles cellulaires, et entraîne à terme une sénescence cellulaire [38]. Une déficience en télomérase chez les humains est associée à un développement prématuré de maladies (fibrose pulmonaire, dyskératose congénitale, et anémie aplastique) qui implique un défaut de capacité régénératrice des différents tissus [39].
Plusieurs mécanismes interviennent dans la régulation de la reproduction des télomères. La shelterine est une protéine complexe qui fixe les télomères et régule la reproduction [40] [41]. Un défaut de fonctionnement de la formule pour les composants de la shelterine entraîne un déclin rapide de la capacité régénératrice des tissus, et un vieillissement accéléré. Les sirtuines sont suspectées de jouer un rôle dans la maintenance des télomères [43] via des modulations épigénétiques. La protéine SIRT-6 module la chromatine télomérique [44] et le cycle cellulaire [45].
Des télomères très courts pourraient activer une voie de réponse des lésions de l’ADN impliquant les protéines p53 et p21WAF1 dans les cellules vieillissantes [46]. La fixation et l’activité transcriptionnelle de la protéine p53 de l’ADN augmentent avec l’âge, et la protéine p21WAF1 qui favorise l’activité des cellules sénescentes dépend de la protéine p53. De plus, la protéine PARP pourrait être impliquée dans l’activation post-traductionnelle de p53 dans les cellules vieillissantes : la protéine peut être associée à la PARP, et l’inhibition de l’activité de la PARP conduit à la suppression de l’expression de p21 en réponse aux lésions de l’ADN [46]. L’activation de la protéine PARP est associée à une érosion accélérée des télomères et à une sénescence avancée [47].
Le rétrécissement des télomères est souvent décrit comme une horloge biologique des cellules somatiques, provoquant ainsi une sénescence cellulaire. Ceci ressemble à une stratégie anti-cancer supplémentaire, car elle empêche la prolifération incessante des cellules. Ce mécanisme pourrait ainsi favoriser la survie pendant la jeunesse en réduisant le développement de cellules cancéreuses, mais il pourrait aussi à terme remettre en question l’homéostasie tissulaire en provoquant une sénescence excessive. Bien que cela soit parfois interprété comme un mécanisme de sénescence programmée (voir «sénescence cellulaire »), son rôle dans le vieillissement des phénotypes n’a pas été clairement établi.
L’épigénétique comme facteur clé du controle de la durée de vie
Mécanismes épigénétiques. Source : http://en.wikipedia.org/wiki/Epigenetics
L’épigénétique est au cœur du processus du vieillissement car elle décrit des changements d’activité cellulaire dans le temps via une modulation des voies traductionnelles. Les altérations épigénétiques sont provoquées par un système enzymatique complexe qui garantit l’entretien et l’évolution des configurations épigénétiques. Elles sont liées à l’activation des gènes inflammatoires et au développement des maladies liées au vieillissement (le cancer, la démence et l’athérosclérose).
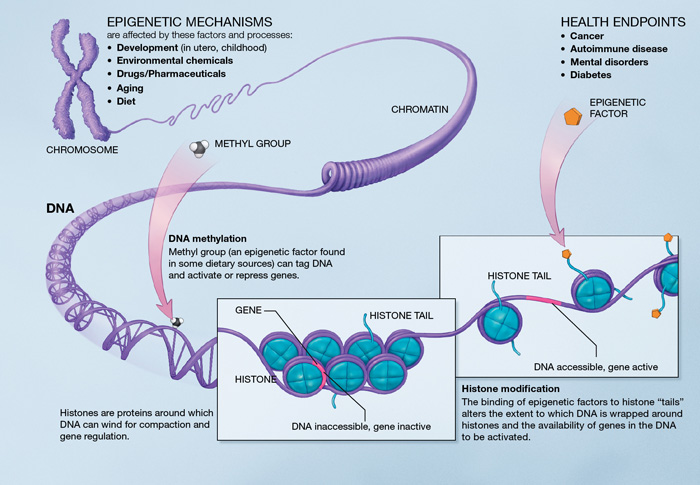
La méthylation de l’ADN. Il existe une hypo ou hyperméthylation liée à l’âge de certains locus de l’ADN [48]. Sur un promoteur d’un gène, la méthylation agit de manière à repousser la transcription génétique. L’hypométhylation pourrait augmenter le taux de mutation de l’ADN. Les gènes hyperméthylés associés à l’âge comprennent des gènes qui encodent un récepteur d’oestrogènes, un facteur de croissance insulinoïde de type 2 (IGF-2) et un suppresseur tumoral [48]. Les modifications de la méthylation des configurations de l’ADN peuvent particulièrement affecter les cellules souches [49]. Cependant, il n’y a pas de preuve que la durée de vie puisse être prolongée par une modification des configurations de méthylation de l’ADN.
Le remodelage de la chromatine. Avec l’âge, le niveau des facteurs de remodelage de la chromatine diminue [49] [50], et on constate une perte ou une redistribution générale de l’hétérochromatine. Des études ont découvert la preuve d’un lien entre la formation d’hétérochromatine et une stabilité chromosomique. Les extrémités chromosomiques sont assemblées dans des zones de l’hétérochromatine [52] [53] de façon à ce que les altérations épigénétiques puissent influencer la régulation de la longueur des télomères.
Modifications des histones. Supprimer certains composants des complexes de méthylation de l’histone augmente la durée de vie chez les invertébrés [54] [56]. L’inhibition de l’histone déméthylase chez les vers pourrait augmenter la durée de vie en ciblant les voies clés de la longévité. Les sirtuines SIRT-1, 6 et 7 (2) jouent un rôle clé dans les modifications des histones. Chez les mammifères, les vertus de la surexpression de la SIRT-1 comprennent une stabilité génomique améliorée. La SIRT-6 régule la stabilité génomique, les signaux NF-κB et l’homéostasie du glucose via la désacétylation de l’histone [59] [60] [61].
Altérations transcriptionnelles. Une augmentation associée à l’âge dans le bruit transcriptionnel [62] entraîne une surproduction et une maturation des ARNm [63] [64] et des miARN (voir Une approche systémique du vieillissement – miARN). Des changements transcriptionnels pour l’encodage de composants inflammatoires, des voies de dégradation des mitochondries et des lysosomes [65] montrent encore une fois la façon dont les différentes caractéristiques peuvent être entremêlées.
Le vieillissement et la détérioration de la réponse des protéines dépliées
Le vieillissement et de nombreuses maladies qui y sont liées (la maladie d’Alzheimer et la dégénérescence maculaire liée à l’âge), ont été associés à l’accumulation cellulaire et à un regroupement de protéines dépliées [66] qui provoquent ainsi un dysfonctionnement cellulaire. L’homéostasie des protéines est régulée par des mécanismes intra-cellulaires complexes. En réponse à ce stress, ces mécanismes sont adaptés pour préserver le fonctionnement du protéome.
Les protéines chaperonnes jouent un rôle majeur dans le pliage, l’assemblement, le transport et le ciblage par dégradation des protéines. Les changements liés au vieillissement remettent en cause leur activité et leur synthèse [68] et entraînent des problèmes dans le contrôle de qualité des protéines. Un approvisionnement d’ATP endommagés faisant partie de vieux organismes (voir « un dysfonctionnement des mitochondries ») peut perturber la fonction des chaperons. Des modifications au sein de la protéine substrat (par exemple l’accumulation de produits finaux de glycation avancés via des modifications non-enzymatique [69]) peuvent perturber la capacité d’une protéine chaperonne à identifier sa cible.
Les manipulations pharmacologiques et génétiques qui augmentent la durée de vie chez les organismes modèles stimulent souvent l’autophagie (5) [70] [71]. Les inhibiteurs de l’autophagie compromettent les effets positifs de l’activation des CR, SIRT-1, l’inhibition des IIS et l’administration de rapamycine, de resvératrol ou encore de spermidine [71] [72].
De nombreuses voies intégrées co-régulent la dégradation des protéines. Le vieillissement a un effet négatif sur les interférences protéines [73] [74]. La déficience des voies de signalisation liées au vieillissement (voie de facteur de croissance insulinoïde 1 (IGF-1), qui assure une interférence entre la protéasome et l’autophagie (5)
L’entretien de la protéose des mitochondries est un véritable défi en raison des niveaux des espèces ROS dans cet organite. La façon la plus commune d’éliminer le dysfonctionnement des mitochondries pourrait être la dégradation autophagique [75], connue sous le nom de mitophagie. Cependant, de nombreuses études ont mentionné un dysfonctionnement du nettoyage des mitochondries et des troubles liés au vieillissement [76]. La réponse mitochondriale des protéines dépliées (UPRmt) est activée lorsque les protéines dépliées et mal dépliées s’accumulent dans les mitochondries [77] [78]. Des études indiquent que la communication entre les mitochondries et le noyau peuvent provoquer l’expression des gènes nucléaires qui encodent les protéines mitochondriales en réponse aux protéostases altérées [79] [80]. Les échecs de communication entre les mitochondries et le noyau pourraient par conséquent être impliqués dans l’affaiblissement de la protéostasie mitochondriale (voir « Le dysfonctionnement mitochondrial »).
La sirtuine mitochondriale SIRT-3 (2), une protéine déacylase, qui influence presque tous les aspects de la biologie mitochondriale (comprenant l’oxydation de nutriments, une production d’ATP, une détoxification des ROS, des dynamiques mitochondriales mtUPR) [81]. Une contrainte protéotoxique provoquée par un manque de protéostasie des mitochondries active la SIRT-3, et les mécanismes antioxydants de défense des mitochondries [82]. L’expression de la SIRT-7, un composant de la branche régulatrice du mtUPR, est réduit chez les cellules souches hématopoïétiques, et sa surexpression dans ce réglage peut améliorer la capacité régénératrice de ces cellules [83].
Vieillissement et défaillance des mitochondries
La théorie du stress oxydatif (autrement dite théorie des radicaux libres) affirme que le dysfonctionnement de cellules souches mitonchondriales liées au vieillissement est causé par des niveaux élevés de dérivés réactifs de l’oxygène (DRO) chez les mitochondries (4) [84]. Cependant, le rôle de la contrainte oxydative est susceptible d’avoir été mal compris au cours des premières phases de recherche sur le vieillissement. De récentes expériences ont montré que des niveaux élevés de DRO pourraient prolonger la durée de vie [85] [86] [87], alors cela n’a pas été le cas pour la défense antioxydante améliorée. De plus, il a été démontré que les dérivés réactifs de l’oxygène (DRO) déclenchent la prolifération et les réactions de survie dans des conditions de stress [89]. Le mécanisme exact entre le DRO et les pathologies liées au vieillissement provoquées par le stress oxydatif n’est pas encore clair. Ils pourraient stimuler par exemple l’autophagie (5).
Le DRO pourrait être considéré comme un signal de survie provoqué par le stress. Lors du vieillissement, le stress cellulaire et le nombre de lésions augmentent. Les niveaux de DRO augmentent parallèlement dans le but de maintenir la survie. Au-delà d’un certain point, ces niveaux trahissent leur devoir homéostatique et pourraient possiblement aggraver les lésions associées à l’âge [92].
Le dysfonctionnement des mitochondries semble être une caractéristique du vieillissement. De façon remarquable, la voie métabolique de l’OXPHOS (phosphorylation oxydative) des mitochondries est perturbée au cours de vieillissement avant toute lésion significative du mtADN [93] : cela suggère que les lésions du mtADN seules ne peuvent pas expliquer le dysfonctionnement des mitochondries. D’autres causes ont alors été étudiées. Les communications centrales des mitochondries sont nécessaires à la biogenèse de celles-ci et à leur réaction aux voies de stress. [94]. Des altérations de ces voies de communications pourraient provoquer un dysfonctionnement des mitochondries.
Les sirtuines (2) se désactivent au fur à mesure que l’âge chronologique avance. La SIRT-1 et 3 régulent négativement le HIF-1α (6) [93] [95] [96]. La modulation négative du HIF-1α entraîne une production de facteur de transcription mitochondrial A (mtTFA) est un activateur clé de la transcription mitochondriale. L’appauvrissement du pool NAD+ (1) pourrait ainsi expliquer la dérégulation de l’expression des mitochondries et la perturbation de l’OXPHOS [93].
Les sirtuines (2) sont également impliquées dans la régulation des mitochondries via l’activation du PGC-1α (6) [97] [98]. La réparation de l’affection de l’ADN par excision de nucléotides entraîne des anomalies au niveau des mitochondries qui seraient causées par une activation décroissante de l’axe NAD/SIRT-1/PGC-1α au moyen d’une hyperactivation des capteurs de lésions de l’ADN PARP-1 [99] ( voir « les Lésions du Génome » ). L’expression de la protéine (3) p53 peut également réprimer le PGC-1α (récepteur gamma activé par les proliférateurs de peroxysomes) et déréguler la fonction des mitochondries comme conséquence de l’attrition télomérique par exemple [100].
Quant à savoir si un dysfonctionnement associé à l’âge apparaît par l’intermédiaire d’une voie de PGC-1α -dépendant et du HIF-1a, ou une combinaison des deux, rien n’est encore clair.
Le dysfonctionnement (4) des mitochondries pourrait être provoqué par une altération de la communication entre le noyau et les mitochondries à travers une dérégulation de l’activité (2) des sirtuines mitochondriales. Elle a de nombreux effets négatifs sur le métabolisme des cellules en raison du rôle crucial des mitochondries dans le métabolisme énergique cellulaire. Par exemple, cela pourrait conduire à une mauvaise gestion de l’apoptose et ainsi provoquer des réactions inflammatoires [101].
Détection de nutriments : les vertus de la restriction calorique pour prolonger la durée de vie
Les voies de détection de nutriments adaptent le comportement cellulaire, le métabolisme, le développement et la longévité aux niveaux de nutriments. L’une d’entre elles, la voie de signalisation (IIS) insuline/ IGF-1, est impliquée dans la détection du glucose et est également liée à la longévité. L’hormone IGF-1 est produite en réponse à l’hormone de croissance (GH). Diverses manipulations génétiques qui affaiblissent l’intensité de la détection à un certain point au sein de la voie (GH, IGF-1, AKT ou FoxO) ont permis d’augmenter la durée de vie chez les vers, les mouches et les souris.
D’autres voies interconnectées de détection des nutriments comprennent les sirtuines (2), et les enzymes AMPK et mTOR. L’AMPK et la SIRT1 indiquent la rareté des nutriments, contrairement au mTOR qui détecte les concentrations élevées en acides aminés. Les sirtuines décèlent des états faibles en énergie en détectant des niveaux élevés en NAD+, et l’AMPK décèle des niveaux d’énergie élevés en détectant des niveaux d’AMP. Par conséquent, leur régulation positive favorise un vieillissement en bonne santé. Les réponses des SIRT1 et de L’AMPK sont coordonnées [106]. L’activation de l’AMPK interrompt le mTORC1 [107]. L’administration de metformine pourrait prolonger la durée de vie, par l’intermédiaire de l’activation de l’AMPK chez les vers et les souris [108] [109] [110]. L’administration de rapamycine prolonge la durée de vie par l’inhibition de mTOR [111]. Puisque la SIRT-1 active le PGC-1α (6) [98], le PGC-1a pourrait établir un lien entre la détection de nutriments et la fonction des mitochondries.
Le jeûne provoque également une baisse d’activité de la protéine PARP [112]. Ceci renforce l’hypothèse que les sirtuines (2) et les protéines PARP sont en « compétition » pour le pool cellulaire NAD+(1).
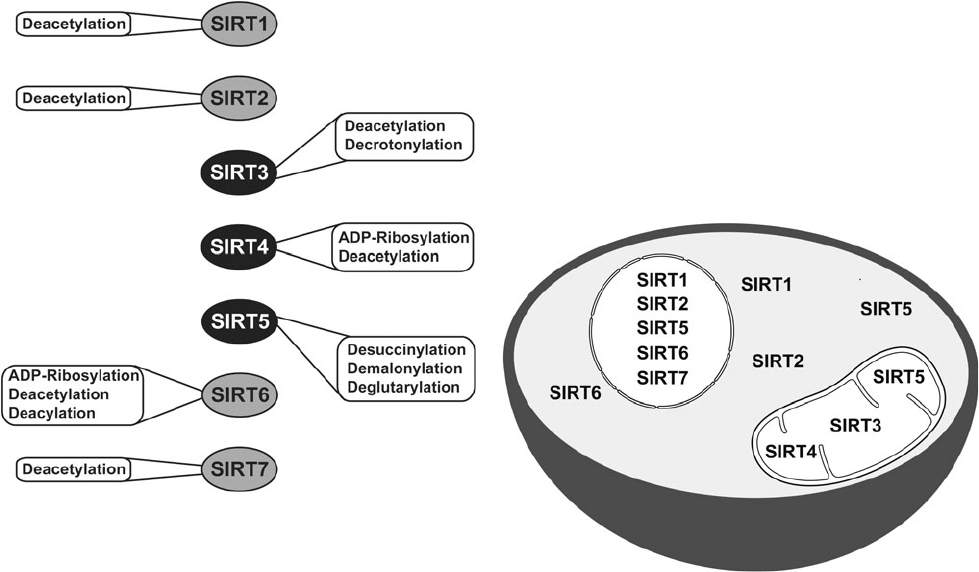
Les activités catalytiques la localisation subcelluaire des sirtuines chez les mammifères. Source : David B. Lombard et Surinder Kumar, Mitochondrial sirtuins and their relationships with metabolic disease and cancer, 2015.
Paradoxalement, un faible taux d’IIS est typique du vieillissement normal des phénotypes, alors qu’une baisse constitutive d’IIS augmente la durée de vie. Un modèle qui comprend une baisse d’IIS constitue une réponse défensive aux lésions systémiques qui peuvent harmoniser ces résultats. Une modulation négative de l’IIS est supposée provoquer une lenteur du métabolisme et de la croissance cellulaire, et ainsi réduire le taux de lésions cellulaires. La dérégulation de la détection des nutriments peut être interprétée comme une réaction de survie de l’organisme, qui à terme se détériore avec l’âge.
Le principe du déterminisme génétique fournit une théorie sous-jacente appropriée pour des modèles de recherche qui évaluent les facteurs nutritionnels participant à la durée de vie. Les études nutritionnelles semblent mieux nous permettre de comprendre la détermination de la longévité que celles sur le vieillissement. [113]
La sénescence cellulaire et le vieillissement phénotypique
Une cellule sénescente arrête de se diviser et comporte des changements phénotypiques notables, comme la sécrétion des molécules pro-inflammatoires [114]. La sénescence cellulaire est un composant crucial du développement de l’organisme et on l’observe à chaque étape de la vie. Elle peut-être causée soit par des lésions de l’ADN (comprenant le rétrécissement des télomères), une détection excessive des mitogènes et des oncogènes, soit par l’expression de l’inhibiteur de cycle cellulaire. Les cellules sénescentes s’accumulent dans certains tissus avec le vieillissement chronologique [115]. De façon plus intéressante, la sénescence peut être provoquée soit par les voies des cellules endogènes soit par les voies de détection extracellulaires.
Deux voies de sénescence essentielles activées en réponse aux altérations des oncogènes ont été identifiées: p16INK4a/Rb and p19ARF/p53 (3). Les niveaux de suppresseur de tumeur p16INK4a/ correspondent à l’âge chronologique. Les altérations p16INK4a and p19ARF sont encodées par le même locus génétique (INK4a/ARF) qui est associé au vieillissement, selon des études statistiques.
Une fois de plus, il existe un désaccord en ce qui concerne l’effet du p16INK4a et du p19ARF sur la durée de vie. L’activation de la protéine p53 (3) et du locus INK4a/ARF seraient considérés comme une réaction compensatoire bénéfique afin d’endiguer la prolifération des cellules endommagées. Cependant, lorsque la lésion est généralisée, la capacité régénératrice des tissus peut être épuisée ou saturée, et dans ces conditions extrêmes, les réactions de la protéine p53 et du locus INK4a/ARF peuvent alors accélérer le vieillissement.
La sénescence pourrait être une stratégie visant à empêcher une cellule de proliférer jusqu’au stade de tumeur. Ce système anti-cancéreux requiert un système immunitaire efficace pour régénérer les tissus en phagocytant les cellules sénescentes et les cellules souches robustes. Cependant, avec le vieillissement chronologique, les cellules sénescentes s’accumulent en raison d’un ralentissement du renouvellement cellulaire et des mécanismes immunitaires endommagés qui éliminent les cellules sénescentes. La sénescence est donc censée favoriser la survie aux premiers stades de la vie en empêchant le développement du cancer, mais à terme, elle limite l’espérance de vie à force d’accumulation des cellules sénescentes. Décider si la sénescence cellulaire est un mécanisme de vieillissement programmé nécessite plus de recherches : le lien entre la sénescence cellulaire et le vieillissement phénotypique reste incertain.
Le vieillissement et l’épuisement des cellules souches

En raison de la sénescence des cellules souches, les tissus vieillissants n’arrivent plus à se régénérer aussi facilement que les tissus plus jeunes : ils subissent moins de divisions cellulaires. Par exemple, le vieillissement des cellules hématopoïétiques entraîne une diminution de la production immunitaire adaptative des cellules et est donc impliqué dans l’immunosénescence. Des comportements similaires sont identifiés dans le cerveau, les os, et les fibres musculaires. Les lésions de l’ADN [116] et la surexpression des protéines qui inhibent le cycle cellulaire [117] pourraient expliquer ce comportement.
Bien que les cellules souches adultes expriment la télomérase, elles ne sont pas immunisées contre l’attrition télomérique. [118] [119] L’accumulation de lésions et de mutations non télomériques de l’ADN joue également un rôle dans l’épuisement des cellules souches [120] [121]. Au sein du système hématopoïétique, les mutations qui favorisent la prolifération et la survie sont sélectionnées, et régulièrement, l’augmentation du nombre de cellules souches associée aux mutations précancéreuses sont observées [122] [123] [124] [125]. Les lésions de l’ADN pourraient également diminuer le nombre de cellules souches en leur faisant subir une apoptose ou une sénescence.
Il a été rapporté que la surexpression de la sirtuine SIRT3 améliorerait la capacité régénératrice des vieilles cellules souches hématopoïétiques [126], ce qui suggère que les sirtuines sont aussi impliquées dans l’entretien de la fonction des cellules souches.
L’épuisement des cellules souches est suspecté de jouer un rôle décisif dans le vieillissement de l’organisme, car il freine le renouvellement cellulaire, un composant essentiel dans le processus de vieillissement. Dans une revue datant de 2016, de nombreuses hypothèses décrites par Schultz et Sainclair tentent d’expliquer comment les cellules souches individuelles peuvent fonctionner tout au long de la vie d’un individu. Une de ces hypothèses est le fait que les cellules souches se servent de mécanismes spéciaux pour empêcher ou inverser les lésions ou les changements épigénétiques, qui comprennent l’entretien de longs télomères et d’une amélioration de la protéostasie. Une autre hypothèse serait que certaines cellules souches se dégradent avec l’âge, tandis que les plus saines sont sélectionnées. Les modèles de fonction améliorée des cellules souches entraînent une amélioration constante de la fonction tissulaire avec ce que l’on pourrait prédire si le vieillissement des cellules souches est un élément moteur du vieillissement des tissus et de l’organisme [128] [129]. Comprendre l’épuisement des cellules souches suscite de nombreuses répercussions sur la médecine régénératrice.
Une approche systémique du vieillissement et de la détermination de la durée de vie
En 2014, des scientifiques ont démontré que le sang jeune pouvait rajeunir de vieux tissus en combinant ces deux éléments chez les souris [130]. De plus, les manipulations qui prolongent la durée de vie ciblent un tissu qui pourrait avoir des conséquences bénéfiques sur d’autres [131] [132] [133]. Ces résultats intéressants ont entraîné une recherche active sur les facteurs systémiques circulants, qui pourraient contrôler et coordonner le taux de vieillissement des différents tissus et organes.
L’inflammation chronique liée au vieillissement
« L’inflammaging » est un phénotype pro-inflammatoire observé chez les mammifères vieillissants [134]. Les cytokines jouent un rôle majeur dans la réaction inflammatoire, elles permettent la communication entre les cellules immunitaires et les cellules somatiques. Leur réponse chronique pourrait être partiellement responsable de la vulnérabilité, le développement des maladies liées au vieillissement, et le déclin du vieil âge [135] [136].
De multiples causes sont examinées : l’accumulation de lésions dans les tissus pro-inflammatoires au fil du temps, la sécrétion de cytokines pro-inflammatoires par des cellules sénescentes, le système immunitaire défaillant qui ne parvient pas à éliminer les éléments pathogènes et les cellules-hôtes défaillantes, la mauvaise gestion de l’apoptose en raison du dysfonctionnement des mitochondries, une mauvaise réponse de l’autophagie et une accumulation des protéines endommagées qui entraîne une réaction inflammatoire, un changement des niveaux d’hormones qui régulent la production de cytokine (dont la testostérone) et l’activation améliorée du facteur de transcription NF-κB.
Ces altérations donnent lieu à une amélioration de la production d’interleukine 1β (IL-1β), un facteur de nécrose tumorale [101] [134] qui entraîne une libération du TNF-α. L’interleukine-6 (IL-6) est libérée par le TNF-α et l’interleukine IL-1β.
Le facteur nucléaire –κB (NF-κB) participe à la réponse immunitaire et à la réponse au stress. Il contrôle de nombreux gènes liés à l’inflammation et est considéré comme un responsable moléculaire de l’inflammaging. L’inhibition de NF-κB prévient des caractéristiques du vieillissement avancé chez différentes souris modèles [137] [138]. De plus, l’inhibition de la protéine NF-κB entraîne un rajeunissement phénotypique des tissus [139]. Il faut noter que l’expression de la sirtuine SIRT-1 inhibe le NF-κB.
Les sirtuines pourraient avoir également une influence sur l’inflammaging. Des études ont révélé qu’en procédant à la désacétylation des histones et des composants des voies de signalisation comme le NF-κB, la sirtuine SIRT1 régule négativement les gènes associés à l’inflammation. La diminution des niveaux de SIRT-1 est associée au développement et à la progression de nombreuses maladies inflammatoires, et l’activation pharmacologique du SIRT1 éviterait des réactions inflammatoires chez les souris [141] [142] [143]. Les sirtuines SIRT-2 et 6 réguleraient négativement la réponse inflammatoire au moyen de la désacétylation des sous-unités du NF-κB et de la répression transcriptionnelle de leurs gènes cibles [60] [144].
Des études sur le facteur de dégradation d’ARNm AUF-1 apportent une preuve in vivo du lien entre le vieillissement et l’inflammation. L’AUF-1 contrôle la dégradation l’ARNm de cytokine et intervient dans la régulation de la limite de la réaction inflammatoire. Les souris qui ont une carence en AUF-1 présentent une sénescence cellulaire prononcée et un vieillissement prématuré des phénotypes. Il faut noter que l’AUF-1 contribue également au maintien de la longueur des télomères [145].
Une fois de plus, l’inflammation liée au vieillissement peut être interprétée comme un mécanisme de défense qui finit par devenir nocif avec le temps. De faibles niveaux de réponse inflammatoire seraient favorables à la réparation et à la régénération tissulaire par l’activation du système immunitaire ; cependant, des niveaux plus élevés pourraient aggraver les lésions. L’inflammation liée au vieillissement pourrait également fragiliser la fonction des cellules souches [146].
![]()
ARNmis
Les micro-ARN (ARNmis) sont un groupe non codant de petits ARNs. Ils peuvent moduler l’expression génétique en ciblant l’ARNm avec une séquence complémentaire. Il faut noter que les ARNmis peuvent contrôler la durée de vie chez les invertébrés [147] [148] [149] en interagissant avec les composants des réseaux de la longévité ou en régulant les capacités régénératrices des cellules souches. Ils pourraient devenir un biomarqueur intéressant du vieillissement et des maladies qui y sont liées, car la caractérisation des ARNmis révèle des altérations considérables dans leurs niveaux d’expression [143].
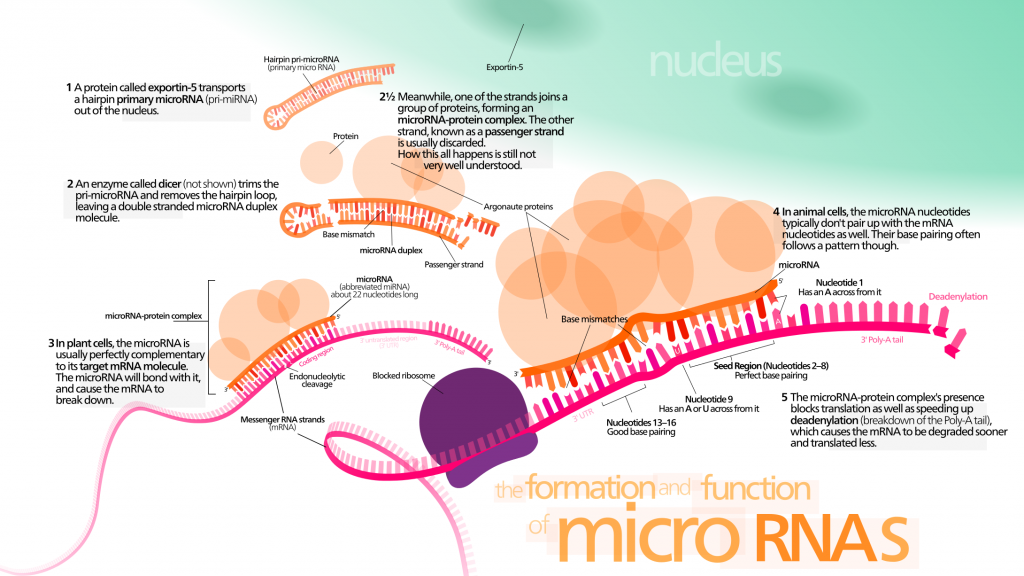
Des ARNmis sans cellules ont attiré l’attention des biologistes, car ils pourraient apporter des informations utiles pour un diagnostic précoce du cancer et d’autres maladies liées au vieillissement. Les ARNmis circulants seraient des facteurs systémiques majeurs du vieillissement. Ils sont connus pour participer à de nombreux aspects du processus de vieillissement :
- La longueur des télomères Les ARNmis participerait à la régulation de la longueur des télomères. De nombreux ANRmis prennent pour cible les ARNm impliqués dans la maintenance des télomères [153]. De plus, chez les humains, l’ANRmi miR-138 est associé négativement au niveau des télomérases reverse transcriptase (hTERT) [154].
- La sénescenceIl existe une preuve qui montre que les ARNmis régulent la sénescence cellulaire et contrôlent l’expression des protéines p53 et p21 [155]. La SIRT-1 pourrait désacétyler et contrôler l’activité de la protéine p53. Le mir -34a est une p53 induite par l’ARNmi, qui peut réguler le niveau d’expression du SIRT-1 chez les souris [156]. Le lien entre la SIRT-1, la p53, et le mir -34 α pourrait représenter un circuit de régulation de la sénescence. En outre, au sein des modèles de sénescence induits par le stress et les oncogènes, la protéine p21 est identifiée comme étant la cible de plusieurs ARNmis [157] [158]. Il faut noter que les niveaux de la famille des miR-106b sont réduits au sein des cellules sénescentes. Le gène mir -106a prend notamment pour cible la protéine p21 ARNm [159] et empêche la sénescence cellulaire, indépendamment du fait que la protéine p53 soit inhibée ou non. D’autres membres de la famille des miR-106b favorisent le cycle cellulaire en prenant pour cible la protéine p21 [158] [160]. Les ARNmis sont suspectés de se trouver au point de croisement entre la sénescence et les maladies liées au vieillissement.
- L’épuisement des cellules souches Certaines ARNmis régulent les propriétés des cellules souches et pourraient ainsi et entretenir la capacité de renouvellement des cellules.
L’inflammation Les ARNmis participerait à la régulation de l’inflammation par l’intermédiaire d’interactions avec la protéine NF-κB [161] [162]. Un petit nombre relatif d’ARNmis semble participer à la régulation de l’inflammation: leurs prototypes sont les micro-ARN155, micro-ARN7 et le micro-ARN 146a [163] [164]. Dans des conditions physiologiques, leur transcription est à un niveau de base, mais les résultats de signalisation pro-inflammatoires ont pour conséquence une forte co-induction de leur expression au moyen d’un mécanisme dépendant de NF-kB.
![]()
L’atrophie thymique et son rôle dans l’immunosénescence
Le thymus est un organe qui soutient le développement et la différenciation des lymphocytes T. Les lymphocytes T jouent un rôle décisif dans le système immunitaire adaptatif, qui permet au corps de répondre spécifiquement aux corps étrangers.
Dans les années 1980, Steinman a démontré que la fonction thymique diminue progressivement dès la première année de la vie [166] et que le thymus s’atrophie au cours du vieillissement. Cette involution a pour conséquence une efficacité diminuée du développement des lymphocytes T, et une émigration amoindrie des lymphocytes T naïfs [168]. De ce fait, le système immunitaire adaptatif se dégrade tandis que le système immunitaire inné se renforce.
Alors que le système immunitaire devient moins efficace avec l’âge, l’incidence de maladies telles que les maladies opportunistes, l’auto-immunité ou le cancer augmente [169] [170]. Selon M. R. Dowling et P. D. Hodgkin, ce comportement visiblement néfaste pourrait avoir été sélectionné pour renforcer la lutte contre les infections et ainsi éviter les réactions contre soi-même. La dégradation associée au vieillissement au sein de la fonction immunitaire pourrait ainsi être un effet secondaire regrettable de ce processus de sélection.
![]()
Le rôle de l’hypothalamus
L’hypothalamus communique avec de nombreux tissus au moyen de réseaux hormonaux et neuronaux. Il coordonne les réactions métaboliques et comportementales aux stimuli nutritionnels et environnementaux chez les mammifères. Il pourrait jouer un rôle majeur dans le vieillissement systémique et dans le contrôle de la longévité [172].
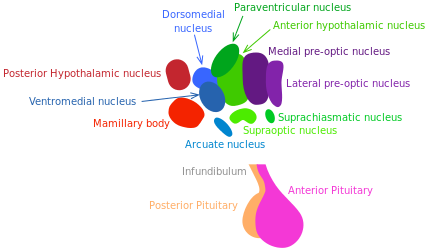
La somatostatine et l’hormone de croissance (GH), un libérateur d’hormones qui sont produites par les neurones hypothalamiques, stimulent et inhibent respectivement la libération de l’hormone de croissance. Il est établi que la voie GH/IGF-1 (régulateur de croissance, réparation tissulaire et métabolique) contrôle la longévité chez les mammifères [173] (voir « Les voies de détection des nutriments »).
Les réactions inflammatoires activent la NF-κB dans l’hypothalamus, déclenchant une voie de signalisation qui réduit la production du GnRH par les neurones. La dégradation de cette hormone pourrait accentuer de nombreux changements systémiques liés au vieillissement comme la fragilité osseuse, la faiblesse musculaire, l’atrophie de la peau, et la neurogénèse amoindrie.
En 2013, Satoh et al. ont démontré que la surexpression transgénique SIRT-1 spécifique au cerveau des souris est liée à un prolongement de la durée de vie. De façon plus intéressante, ces souris présentent des phénotypes cohérents avec un retard du vieillissement [175]. Ces phénotypes révèlent une activité neuronale améliorée, notamment dans le noyau hypothalamique dorso-médian et latéral. Dans cette expérience, des informations complémentaires confirment le rôle du signalement du médiateur SIRT-1 dans ces zones de l’hypothalamus quant au prolongement de l’espérance de vie. Les résultats de Satoh et al. suggèrent que l’activité améliorée de la sirtuine SIRT-1 au sein du noyau hypothalamique latéral et dorso-médian joue un rôle essentiel dans la régulation de la longévité chez les mammifères.
![]()
Notes: des composants de voies de vieillissement intéressants
(1) NAD
Le nicotinamide adenine dinucleotide (NAD) est une coenzyme qui participe à plusieurs réactions de redox cellulaire, dont la synthèse ATP. Il joue un rôle essentiel dans de nombreuses voies métaboliques, et il est à la fois un substrat des protéines PARP et des sirtuines. Sa forme oxydée est connue sous le nom de NAD+, et sa forme réduite est le NADH. La synthèse du NAD+ peut avoir lieu à la fois à partir des acides aminés de novo, ou en recyclant des composants préformés. Le pool cellulaire du NAD+ pourrait s’appauvrir lors du vieillissement [176]. Cela pourrait être dû à la dégradation des voies de signalisation et/ou à une dégradation améliorée (peut-être au moyen d’activation de la protéine PARP). Les niveaux NAD+ sont supposés être une signature importante du vieillissement moléculaire. Les précurseurs du NAD+ ont été soigneusement étudiés, car la promotion de la réplétion du pool de NAD+ pourrait activer les sirtuines et aider à inverser le vieillissement.
(2) Les Sirtuines
Les sirtuines font partie de la famille des protéines dépendantes du NAD+ qui sont supposées jouer un rôle essentiel dans le processus de vieillissement. La première sirtuine à être découverte a été Sir2. Dans les années 1990, Dr Leonard Guarante de MIT a découvert que le gène Sir2 contrôle la longévité chez les levures. Le gène orthologue chez le vers C. Elegans s’est avéré être également essentiel dans le contrôle de la longévité. Aujourd’hui, les sirtuines sont supposées être des facteurs clés de la longévité chez de nombreuses autres espèces, ainsi que chez les humains. Sept d’entre elles ont été identifiées chez les mammifères [177]. Elles ont une activité enzymatique, et elles peuvent également provoquer des changements dans le métabolisme cellulaire et la physiologie. Les sirtuines SIRT-1, 6 et 7 sont des protéines nucléaires, elles ont un impact épigénétique sur l’expression génique en raison d’une désacétylation des histones [178]. La sirtuine SIRT-2 participe au contrôle du cycle cellulaire [178]. Les sirtuines SIRT-3 et 5 régulent les enzymes métaboliques au sein des mitochondries [178]. Elles sont supposées participer à la réaction du corps par rapport à la restriction calorique.
(3) p53
La protéine p53 un suppresseur tumoral connu pour son implication dans le mécanisme contre le cancer. Elle peut activer la réparation des protéines de l’ADN et amorcer une apoptose ou une sénescence en réponse aux lésions de l’ADN. La surexpression de la protéine p53 est délétère, car elle conduit à une sénescence et une apoptose excessive [179]. La protéine p53 peut à la fois inhiber ou stimuler l’autophagie en fonction des conditions de stress [180] [181]. Elle pourrait également avoir un impact sur le vieillissement et le métabolisme, au moyen de son implication dans la fonction des mitochondries [182]. L’activité contraignante et transcriptionnelle de la protéine p53 de l’ADN augmente avec l’âge. La protéine p53 a un rôle ambigu dans le processus de vieillissement, cependant il semble qu’elle régule un équilibre fondamental entre le vieillissement et la suppression tumorale.
(4) Les mitochondries
Les mitochondries sont des organites qui génèrent la majeure partie de l’adénosine triphosphate (ATP) de la cellule, utilisé comme une source de l’énergie chimique. La phosphorylation oxydative (OXPHOS) est la voie métabolique dans laquelle les cellules utilisent des enzymes afin d’oxyder les nutriments et ainsi promouvoir la libération de l’énergie qui sera utilisée pour reformer l’ATP. Cela se produit au sein des mitochondries. Les sirtuines sont réputées pour jouer un rôle dans la régulation de l’activité des mitochondries [178] [185]. La mitophagie est la suppression des mitochondries par autophagie. Elle se produit lors d’une réaction contre les lésions ou le stress. Une mitophagie défaillante (voir « Perte de protéostases ») a été identifiée comme un facteur majeur du dysfonctionnement des mitochondries [186].
(5) L’autophagie
L’autophagie est une voie de régulation responsable de la dégradation des lysosomes des organites cytoplasmiques ou des composants cytosoliques [181]. L’activité autophagique diminue avec l’âge [72] [184], mais elle est régulée pour répondre aux restrictions caloriques. Cette régulation peut être interprétée comme un effort pour augmenter les réserves d’énergie lorsque les nutriments se font rares. Le dérèglement de l’autophagie est un composant fondamental du vieillissement cellulaire, associé au dysfonctionnement mitochondrial et du manque de protéostases.
(6) Les gènes PGC-1α et la protéine HIF-1α
L’activité du gène PGC-1α et de la protéine HIF-1α peut être modulée par des sirtuines [187] [188]. Le facteur de transcription PGC-1α contrôle une réponse métabolique qui comprend la mitochondriogénèse, l’optimisation des défenses anti oxydantes et l’amélioration de l’oxydation des acides gras [189]. La protéine HIF-1α est une sous-unité du facteur de transcription HIF-1. Le facteur de transcription HIF-1 a un rôle important dans la réponse cellulaire au niveau d’oxygène systémique chez les mammifères. Il participe à la prolifération et la survie cellulaire, et à la réaction à l’hypoxie. Le facteur de transcription HIF-1 entraîne également une transcription génique impliquée dans la prolifération et la survie cellulaire, ainsi que le métabolisme du glucose et du fer [191]. Gomes et al. ont émis l’hypothèse que l’AMPK agit comme un commutateur entre la régulation du PGC-1α– dépendant et du HIF-1α-dépendant des mitochondries par les sirtuines. [93].
(7) Les restrictions caloriques
Depuis les années 1930, les restrictions caloriques (RC) sont réputées pour prolonger la longévité chez de nombreuses espèces et les mammifères. Elles sont supposées favoriser le métabolisme oxydatif des mitochondries et la tolérance au stress [177]. Des études ont tendance à montrer que l’effet la RC sur le prolongement de la durée de vie dépend des sirtuines [192] [193]. La théorie de l’évolution peut apporter une connaissance intéressante de ces mécanismes. Lorsque les réserves d’énergie dans l’environnement se font rares, le métabolisme passe sur le mode éco-énergétique. L’objectif est de favoriser la survie, dans l’attente du moment où l’environnement sera plus propice à la reproduction.
Delphine Vendryes
Author
Auteure
Delphine is an engineer and was trained in bio-engineering and in biology at the École Polytechnique X in Paris.
More about the Long Long Life team
Delphine est désormais ingénieure, après une formation en biologie et bio-ingénierie à l’École Polytechnique X à Paris.
En savoir plus sur l’équipe de Long Long Life
Odélie Tacita
Translation
Traduction
Odélie is a trainee specialized translator at Elvesys. She is graduating from the university of Paris-Est Marne-la-Vallée and works in French, English and Spanish.
More about the Long Long Life team
Odélie est apprentie traductrice spécialisée pour Elvesys. Elle termine sa formation à l’université de Paris-Est Marne-la-Vallée et ses langues de travail sont le français, l’anglais et l’espagnol.
En savoir plus sur l’équipe de Long Long Life
![]()
Références
- Finch, C.E.; Kirkwood, T.B. Chance, Development, and Aging; Oxford University Press: New York, NY, USA, 2000.
- Hayflick, L. (2007). Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS genet, 3(12), e220.
- Hayflick, L. Biological aging is no longer an unsolved problem. Ann. N. Y. Acad. Sci. 2007, 1100, 113.
- Hamilton, W.D. The moulding of senescence by natural selection. Theor. Biol. 1966 , 12, 1245.
- Rose, M.R.; Burke, M.K.; Shahrestani, P.; Mueller, L.D. Evolution of ageing since Darwin. Genet. 2008, 87, 363371.
- Lithgow, G.J. Why aging isnt regulated: A lamentation on the use of language in aging literature. Gerontol. 2006, 41, 890893.
- Medawar, P. B. (1952). An unsolved problem of biology.
- Lambert F.L. (2007). Entropy and the second law of thermodynamics.
- Williams, G.C. (1957) Pleiotropy, natural selection and the evolution of senescence. Evolution11, 398411.
- Kirkwood, T. B. (2002). Evolution of ageing. Mechanisms of ageing and development, 123(7), 737-745.
- Wilson, D. L. (1974). The programmed theory of aging. Theoretical aspects of aging, 11-21.
- Lo´pez-Ot´ın, C., Blasco, M. A., Partridge, L., Serrano, M., and G. Kroemer, G. (2013). The Hallmarks of Aging, Cell 153
- Hoeijmakers, J.H. (2009). DNA damage, aging, and cancer. Engl. J. Med. 361 , 14751485
- Gregg, S.Q., Guti´errez, V., Robinson, A.R., Woodell, T., Nakao, A., Ross, M.A., Michalopoulos, G.K., Rigatti, L., Rothermel, C.E., Kamileri, I., et al. (2012). A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology 55 , 609621.
- Murga, M., Bunting, S., Montan˜a, M.F., Soria, R., Mulero, F., Can˜amero, M., Lee, Y., McKinnon, P.J., Nussenzweig, A., and Fernandez-Capetillo, O. (2009). A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Genet. 41, 891898.
- Wallace, D.C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Rev. Genet. 39, 359407.
- Kujoth, G.C., Hiona, A., Pugh, T.D., Someya, S., Panzer, K., Wohlgemuth, S.E., Hofer, T., Seo, A.Y., Sullivan, R., Jobling, W.A., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481484.
- Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J.N., Rovio, A.T., Bruder, C.E., Bohlooly-Y, M., Gidlo¨f, S., Oldfors, A., Wibom, R., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417423.
- Vermulst, M., Wanagat, J., Kujoth, G.C., Bielas, J.H., Rabinovitch, P.S., Prolla, T.A., and Loeb, L.A. (2008). DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Genet. 40, 392394.
- Herbener G. A morphometric study of age-dependent changes in mitochondrial populations of mouse liver and heart. J Gerontol. 1976;31(1):812.
- Stocco DM, Hutson JC. Quantitation of mitochondrial DNA and protein in the liver of Fischer 344 rats during aging. J Gerontol. 1978;33(6):802809.
- Yen T-C, Chen Y-S, King K-L, Yeh S-H, Wei Y-H. Liver mitochondrial respiratory functions decline with age.
- Tauchi H, Sato T. Age changes in size and number of mitochondria of human hepatic cells. J Gerontol. 1968;23(4):454461.
- Stocco DM, Cascarano J, Wilson MA. Quantitation of mitochondrial DNA, RNA, and protein in starved and starved-refed rat liver. J Cell Physiol. 1977;90(2):295306.
- Chinnery, P. F., Samuels, D. C., Elson, J., Turnbull, D. M. (2002). Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism?. The Lancet, 360(9342), 1323-1325.
- Moskalev, A.A., Shaposhnikov, M.V., Plyusnina, E.N., Zhavoronkov, A., Budovsky, A., Yanai, H., and Fraifeld, V.E. (2012). The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev.
- Faggioli, F., Wang, T., Vijg, J., and Montagna, C. (2012). Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Mol. Genet. 21, 52465253.
- Forsberg, L.A., Rasi, C., Razzaghian, H.R., Pakalapati, G., Waite, L., Thilbeault, K.S., Ronowicz, A., Wineinger, N.E., Tiwari, H.K., Boomsma, D., et al. (2012). Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 90, 217228.
- Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shumaker, D.K., Solimando, L., and Goldman, R.D. (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832853.
- Mouchiroud, L., Houtkooper, R.H., Moullan, N., Katsyuba, E., Ryu, D., Canto´, C., Mottis, A., Jo, Y.S., Viswanathan, M., Schoonjans, K., Guarente, L., Auwerx, J. (2013) The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Jul 18;154(2):430-41. doi: 10.1016 /j.cell. 2013.06.016.
- Bai, P., Cant´o, C. (2012). The Role of PARP-1 and PARP-2 Enzymes in Metabolic Regulation and Disease Cell Metabolism 16, September 5
- Braidy, N., Guillemin, G.J., Mansour, H., Chan-Ling, T., Poljak, A., and Grant, R. (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 6, e19194.
- Ying, W., Alano, C.C., Garnier, P., Swanson R.A. (2005). NAD as a Metabolic Link Between DNA Damage and Cell Death. J Neurosci Res. Jan 1-15;79(1-2):216-23
- Campisi, J. (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell, 120(4), 513-522.
- Agarwal, M. L., Taylor, W. R., Chernov, M. V., Chernova, O. B., Stark, G. R. (1998). The p53 network. Journal of Biological Chemistry, 273(1), 1-4.
- Guarente, L. (2014). Linking DNA Damage, NAD+/SIRT1, and Aging. Cell Metabolism 20, November 4.
- Blackburn, E.H., Greider, C.W., and Szostak, J.W. (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Med. 12, 11331138.
- Shay, J. W., Wright, W. E. (2005). Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis, 26(5), 867-874.
- Armanios, M., and Blackburn, E.H. (2012). The telomere syndromes. Nat. Rev. Genet. 13, 693704.
- Palm, W., de Lange, T. (2008). How shelterin protects mammalian telomeres. Rev. Genet. 42, 301334.
- de Lange T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19: 2100-2110.
- Mart´ınez, P., and Blasco, M.A. (2010). Role of shelterin in cancer and aging. Aging Cell 9, 653666.
- Jia, G., Su, L., Singhal, S., Liu, X. (2012). Emerging roles of SIRT6 on telomere maintenance, DNA repair, metabolism and mammalian aging. Mol Cell Biochem. May;364(1-2):345-50. doi: 10.1007/s11010-012-1236-8
- Michishita, E., McCord, R.A., Elisabeth Berber, E., Mitomu Kioi, M., Padilla-Nash, H., Damian, M., Cheung, P., Kusumoto, R., Kawahara, T.L.A., Barrett, J.C., Chang, H.Y., Bohr, V.A., Ried, T., Gozani, O., Chua, K.F. (2008). SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. Mar 27;452(7186):492-6.
- Michishita, E., McCord, R.A., Boxer, L.D., Barber, M.F., Hong, T., Gozani, O., Chua, K.F. (2009). Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6.
- Herbig, U., Jobling, W. A., Chen, B. P., Chen, D. J., Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21 CIP1, but not p16 INK4a. Molecular cell, 14(4), 501-513.
- Vaziri, H., West, M.D., Allsopp, R.C., Davison, T.S., Wu, Y.S., Arrowsmith, C.H., Poirier, G.G., Benchimol, S. (1997). ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. Oct 1;16(19):6018-33.
- Maegawa, S., Hinkal, G., Kim, H.S., Shen, L., Zhang, L., Zhang, J., Zhang, N., Liang, S., Donehower, L.A., and Issa, J.P. (2010). Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 20, 332340.
- Pollina, E.A., and Brunet, A. (2011). Epigenetic regulation of aging stem cells. Oncogene 30, 31053126.
- Pegoraro, G., Kubben, N., Wickert, U., Go¨hler, H., Hoffmann, K., and Misteli, T. (2009). Ageing-related chromatin defects through loss of the NURD complex. Cell Biol. 11, 12611267.
- Schotta, G., Lachner, M., Sarma, K., Ebert, A., Sengupta, R., Reuter, G., Reinberg, D., and Jenuwein, T. (2004). A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 12511262.
- Blasco, M.A. (2007). The epigenetic regulation of mammalian telomeres. Rev. Genet. 8, 299309.
- Gonzalo, S., Jaco, I., Fraga, M.F., Chen, T., Li, E., Esteller, M., and Blasco, M.A. (2006). DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Cell Biol. 8, 416424.
- Greer, E.L., Maures, T.J., Hauswirth, A.G., Green, E.M., Leeman, D.S., Maro, G.S., Han, S., Banko, M.R., Gozani, O., and Brunet, A. (2010). Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383387.
- Siebold, A.P., Banerjee, R., Tie, F., Kiss, D.L., Moskowitz, J., and Harte, P.J. (2010). Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Natl. Acad. Sci. USA 107, 169174.
- Jin, C., Li, J., Green, C.D., Yu, X., Tang, X., Han, D., Xian, B., Wang, D., Huang, X., Cao, X., et al. (2011). Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 14, 161172.
- Oberdoerffer, P., Michan, S., McVay, M., Mostoslavsky, R., Vann, J., Park, S.K., Hartlerode, A., Stegmuller, J., Hafner, A., Loerch, P., et al. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907918.
- Wang, R.H., Sengupta, K., Li, C., Kim, H.S., Cao, L., Xiao, C., Kim, S., Xu, X., Zheng, Y., Chilton, B., et al. (2008). Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312323.
- Kanfi, Y., Peshti, V., Gil, R., Naiman, S., Nahum, L., Levin, E., Kronfeld-Schor, N., and Cohen, H.Y. (2010). SIRT6 protects against pathological damage caused by dietinduced obesity. Aging Cell 9, 162173.
- Kawahara, T.L., Michishita, E., Adler, A.S., Damian, M., Berber, E., Lin, M., McCord, R.A., Ongaigui, K.C., Boxer, L.D., Chang, H.Y., and Chua, K.F. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 6274.
- Zhong, L., DUrso, A., Toiber, D., Sebastian, C., Henry, R.E., Vadysirisack, D.D., Guimaraes, A., Marinelli, B., Wikstrom, J.D., Nir, T., et al. (2010). The histone deacetylase Sirt6 regulates glucose homeostasis via HIF1α. Cell 140, 280293.
- Bahar, R., Hartmann, C.H., Rodriguez, K.A., Denny, A.D., Busuttil, R.A., Doll, M.E., Calder, R.B., Chisholm, G.B., Pollock, B.H., Klein, C.A., and Vijg, J. (2006). Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441, 10111014.
- Harries, L.W., Hernandez, D., Henley, W., Wood, A.R., Holly, A.C., BradleySmith, R.M., Yaghootkar, H., Dutta, A., Murray, A., Frayling, T.M., et al. (2011). Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 10, 868878.
- Nicholas, A., de Magalhaes, J.P., Kraytsberg, Y., Richfield, E.K., Levanon, E.Y., and Khrapko, K. (2010). Age-related gene-specific changes of A-to-I mRNA editing in the human brain. Ageing Dev. 131, 445447.
- de Magalha˜es, J.P., Curado, J., and Church, G.M. (2009). Meta-analysis of agerelated gene expression profiles identifies common signatures of aging. Bioinformatics 25, 875881.
- Kaushik, S., Cuervo, A.M. (2015). Proteostasis and aging. Nature Medicine 21, 14061415
- Feldman, D.E., Frydman, J. Protein folding in vivo: the importance of molecular chaperones. Opin. Struct. Biol. 10, 2633 (2000).
- Calderwood, S.K., Murshid, A., and Prince, T. (2009). The shock of aging: molecular chaperones and the heat shock response in longevity and aginga mini-review. Gerontology 55, 550558.
- Vanhooren, V., Navarrete Santos, A., Voutetakis, K., Petropoulos, I., Libert, C., Simm, A., Gonos, E.S., Friguet, B. (2015). Protein modification and maintenance systems as biomarkers of ageing. Mech Ageing Dev. Nov;151:71-84.
- Morselli, E., Galluzzi, L., Kepp, O., Criollo, A., Maiuri, M. C., Tavernarakis, N., … Kroemer, G. (2009). Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging, 1(12), 961-970.
- Morselli, E., Maiuri, M.C., Markai, M., Megalou, E., Pasparaki, A., Paliaras, K., Galluzzi, L., Criollo, A., Malik, S.A., Madeo, F., et al. (2010). Caloric restriction and resveratrol prolong longevity via the sirtuin-1 dependent induction of autophagy. Cell Death Disease 1, e10.
- Rubinsztein, D.C., Marino, G., Kroemer, G. (2011) Autophagy and aging. Cell 146 , 682695.
- Gavil´an, E., Pintado, C., Gavilan, M.P., Daza, P., S´anchez-Aguayo, I., Castan˜o, A., Ruano, D. (2015). Age-related dysfunctions of the autophagy lysosomal pathway in hippocampal pyramidal neurons under proteasome stress. Aging 36, 19531963.
- Schneider, J.L., Villarroya, J., Diaz-Carretero, A., Patel, B., Urbanska, A.M., Thi, M.M., Villarroya, F., Santambrogio, L., Cuervo, A.M. (2015). Loss of hepatic chaperonemediated autophagy accelerates proteostasis failure in aging. Aging Cell 14, 249264.
- Lemasters, J.J. (2014). Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2, 749754.
- Jensen, M.B., Jasper, H. (2014). Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 20, 214225.
- Heo, J.M., Livnat-Levanon, N., Taylor, E.B., Jones, K.T., Dephoure, N., Ring, J., Xie, J., Brodsky, J.L., Madeo, F., Gygi, S.P., Ashrafi, K., Glickman, M.H., Rutter, J. (2010). A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465480.
- Braun, R.J., Sommer, C., Leibiger, C., Gentier, R.J., Dumit, V.I., Paduch, K., Eisenberg, T., Habernig, L., Trausinger, G., Magnes, C., Pieber, T., Sinner, F., Dengjel, J., van Leeuwen, F.W., Kroemer, G., Madeo, F. (2015). Accumulation of basic amino acids at mitochondria dictates the cytotoxicity of aberrant ubiquitin. Cell Rep. 10, 15571571.
- Jovaisaite, V., Auwerx, J. (2015). The mitochondrial unfolded protein response synchronizing genomes. Curr. Opin. Cell Biol. 33, 7481.
- Haynes, C.M., Ron, D. (2010). The mitochondrial UPR – protecting organelle protein homeostasis. Cell Sci. 123, 38493855.
- McDonnell, E., Peterson, B.S., Bomze, H.M., Hirschey, M.D. (2015). SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. 26, 486492.
- Papa, L., Germain, D. (2014). SirT3 Regulates the Mitochondrial Unfolded Protein Response. Mol Cell Biol. Feb;34(4):699-710.
- Mohrin M., Shin J., Liu Y., Brown K., Luo H., Xi Y., Haynes C.M., Chen D. (2015). Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 13741377.
- Harman, D. (1965). The free radical theory of aging : effect of age on serum copper levels, J. Gerontol 20, 151-153
- Doonan, R., McElwee, J.J., Matthijssens, F., Walker, G.A., Houthoofd, K., Back, P., Matscheski, A., Vanfleteren, J.R., and Gems, D. (2008). Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 22, 32363241.
- Mesquita, A., Weinberger, M., Silva, A., Sampaio-Marques, B., Almeida, B., Le˜ao, C., Costa, V., Rodrigues, F., Burhans, W.C., and Ludovico, P. (2010). Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Natl. Acad. Sci. USA 107, 1512315128.
- Van Raamsdonk, J.M., and Hekimi, S. (2009). Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000361
- Prez, V.I., Bokov, A., Van Remmen, H., Mele, J., Ran, Q., Ikeno, Y., Richardson, A., (2009). Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta, 1790, pp. 10051014
- Sena, L.A., and Chandel, N.S. (2012). Physiological roles of mitochondrial reactive oxygen species. Cell 48, 158167.
- Bellot, G. L., Liu, D., Pervaiz, S. (2013). ROS, autophagy, mitochondria and cancer: Ras, the hidden master?. Mitochondrion, 13(3), 155-162.
- Pole, A., Dimri, M., Dimri, G. P. (2016). Oxidative stress, cellular senescence and ageing. AIMS Molecular Science, 3(3).
- Hekimi, S., Lapointe, J., and Wen, Y. (2011). Taking a good look at free radicals in the aging process. Trends Cell Biol. 21, 569576
- Ana P. Gomes, Nathan L. Price, Alvin J.Y. Ling, Javid J. Moslehi, Magdalene K. Montgomery, Luis Rajman, James P. White, Joo S. Teodoro, Christiane D. Wrann, Basil P. Hubbard, Evi M. Mercken, Carlos M. Palmeira, Rafael de Cabo, Anabela P. Rolo, Nigel Turner, Eric L. Bell, David A. Sinclair (2013). Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell. Dec 19;155(7):1624-38.
- Ryan, M.T., Hoogenraad, N.J. (2007). Mitochondrial-Nuclear Communications. Annu. Rev. Biochem. 76:70122
- Lim, J.H., Lee, Y.M., Chun, Y.S., Chen, J., Kim, J.E., and Park, J.W. (2010). Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxiainducible factor 1alpha. Cell 38, 864878.
- Finley, L. W., Carracedo, A., Lee, J., Souza, A., Egia, A., Zhang, J., …, Pandolfi, P. P. (2011). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α Cancer cell, 19(3), 416-428.
- Gerhart-Hines, Z., Rodgers, J.T., Bare, O., Lerin, C., Kim, S.H., Mostoslavsky, R., Alt, F.W., Wu, Z., and Puigserver, P. (2007). Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 26, 19131923.
- Rodgers, J.T., Lerin, C., Haas, W., Gygi, S.P., Spiegelman, B.M., and Puigserver, P. (2005). Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113118.
- Fang, E.F., Scheibye-Knudsen, M., Brace, L.E., Kassahun, H., Tanima SenGupta, Nilsen, H., Mitchell, J.R., Croteau, D.L., Bohr, V.A. (2014). Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 157, 882896, May 8.
- Sahin, E., and DePinho, R.A. (2012). Axis of ageing: telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 13, 397404.
- Green, D.R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagyinflammation-cell death axis in organismal aging. Science 333, 1109 1112.
- Fontana, L., Partridge, L., and Longo, V.D. (2010). Extending healthy life spanfrom yeast to humans. Science 328, 321326
- Kenyon, C.J. (2010). The genetics of ageing. Nature 464, 504512.
- Slack, C., Giannakou, M.E., Foley, A., Goss, M., and Partridge, L. (2011). dFOXOindependent effects of reduced insulin-like signaling in Drosophila. Aging Cell 10 , 735748.
- Salih, D.A.M, Brunet, A. (2008). FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. Apr; 20(2): 126136.
- Price, N.L., Gomes, A.P., Ling, A.J., Duarte, F.V., Martin-Montalvo, A., North, B.J., Agarwal, B., Ye, L., Ramadori, G., Teodoro, J.S., et al. (2012). SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15, 675690.
- Alers, S., Lo¨ffler, A.S., Wesselborg, S., and Stork, B. (2012). Role of AMPKmTORUlk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 211.
- Anisimov, V.N., Berstein, L.M., Popovich, I.G., Zabezhinski, M.A., Egormin, P.A., Piskunova, T.S., Semenchenko, A.V., Tyndyk, M.L., Yurova, M.N., Kovalenko, I.G., and Poroshina, T.E. (2011). If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (Albany NY) 3, 148157.
- Mair, W., Morantte, I., Rodrigues, A.P., Manning, G., Montminy, M., Shaw, R.J., and Dillin, A. (2011). Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 470, 404408.
- Onken, B., and Driscoll, M. (2010). Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 5, e8758.
- Harrison, D.E., Strong, R., Sharp, Z.D., Nelson, J.F., Astle, C.M., Flurkey, K., Nadon, N.L., Wilkinson, J.E., Frenkel, K., Carter, C.S., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392395.
- Bai, P., Cant´o, C., Oudart, H., Bruny´anszki, A., Cen, Y., Thomas, C., Yamamoto, H., Huber, A., Kiss, B., Houtkooper, R.H., et al. (2011). Cell Metab. 13, 461468.
- McDonald, R. B., Ruhe, R. C. (2011). Aging and longevity: why knowing the difference is important to nutrition research. Nutrients, 3(3), 274-282.
- Tchkonia, T., Zhu, Y., Van Deursen, J., Campisi, J., Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. The Journal of clinical investigation, 123(3), 966-972.
- Wang, C., Jurk, D., Maddick, M., Nelson, G., Martin-Ruiz, C., and von Zglinicki, T. (2009). DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8, 311323
- Rossi, D.J., Bryder, D., Seita, J., Nussenzweig, A., Hoeijmakers, J., and Weissman, I.L. (2007). Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725729.
- Janzen, V., Forkert, R., Fleming, H.E., Saito, Y., Waring, M.T., Dombkowski, D.M., Cheng, T., DePinho, R.A., Sharpless, N.E., and Scadden, D.T. (2006). Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443, 421426.
- Ferron, S. R., Marques-Torrejon, M., Mira, H., Flores, I., Taylor, K., Blasco, M. A. and Farinas, I. (2009). Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci. 29, 14394-14407.
- Flores, I., Canela, A., Vera, E., Tejera, A., Cotsarelis, G. and Blasco, M. A. (2008). The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 22, 654-667.
- Beerman, I., Bock, C., Garrison, B. S., Smith, Z. D., Gu, H., Meissner, A. and Rossi, J. (2013). Proliferation-dependent alterations of the DNA methylation
- Ru be, C. E., Fricke, A., Widmann, T. A., Fu rst, T., Madry, H., Pfreundschuh, M. and Ru be, C. (2011). Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS ONE 6, e17487.
- Corces-Zimmerman, M. R., Hong, W.-J., Weissman, I. L., Medeiros, B. C. and Majeti, R. (2014). Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 111, 2548-2553
- Genovese, G., K¨ahler, A. K., Handsaker, R. E., Lindberg, J., Rose, S. A., Bakhoum, S. F., Chambert, K., Mick, E., Neale, B. M., Fromer, M. et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 24772487.
- Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., Lindsley, C. R., Mermel, C. H., Burtt, N., Chavez, A. et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488-2498.
- Jan, M., Snyder, T. M., Corces-Zimmerman, M. R., Vyas, P., Weissman, I. L., Quake, S. R. and Majeti, R. (2012). Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 4, 149 ra 118.
- Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin, J., Liu, Y., Zhang, D., Scadden, D.T., and Chen, D. (2013). SIRT3 reverses aging-associated degeneration. Cell Rep. 3, 319327.
- Schultz, M.B., Sinclair, D.A. (2016) When stem cells grow old : phenotypes and mechanism of stem cell aging. Development 143, 3-14.
- Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010 Oct 14;6(10):e1001159.
- Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T Jr, Jones DL, Walker DW. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 2011 Nov 2;14(5):623-34.
- Villeda, S. A., Plambeck, K. E., Middeldorp, J., Castellano, J. M., Mosher, K. I., Luo, J., … Wabl, R. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nature medicine, 20(6), 659-663.
- Lavasani, M., Robinson, A.R., Lu, A., Song, M., Feduska, J.M., Ahani, B., Tilstra, J.S., Feldman, C.H., Robbins, P.D., Niedernhofer, L.J., and Huard, J. (2012). Musclederived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nat. Commun. 3, 608
- Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 7991.
- Toma s-Loba, A., Flores, I., Ferna ndez-Marcos, P.J., Cayuela, M.L., Maraver, A., Tejera, A., Borra s, C., Matheu, A., Klatt, P., Flores, J.M., et al. (2008). Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 135, 609622.
- Salminen, A., Kaarniranta, K., Kauppinen, A. (2012). Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 4, 166175
- Morley, J.E., Baumgartner, R.N. (2004). Cytokine-Related Aging Process Journal of Gerontology: MEDICAL SCIENCES, Vol. 59A, No. 9, 924929
- Ferrucci, L., Harris, T.B., Guralnik, J.M., Tracy, R.P., Corti, M.C., Cohen, H.J., Penninx, B., Pahor, M., Wallace, R., Havlik, R.J., 1999. Serum IL-6 level and the development of disability in older persons. J. Am. Geriatr. Soc. 47, 639646.
- Osorio, F.G., B´arcena, C., Soria-Valles, C., Ramsay, A.J., de Carlos, F., Cobo, J., Fueyo, A., Freije, J.M., and L´opez-Ot´ın, C. (2012). Nuclear lamina defects cause ATMdependent NF-kB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 26, 23112324.
- Tilstra, J.S., Robinson, A.R., Wang, J., Gregg, S.Q., Clauson, C.L., Reay, D.P., Nasto, L.A., St Croix, C.M., Usas, A., Vo, N., et al. (2012). NF-kB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122, 26012612.
- Adler, A.S., Sinha, S., Kawahara, T.L., Zhang, J.Y., Segal, E., and Chang, H.Y. (2007). Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 21, 32443257.
- Xie, J., Zhang, X., and Zhang, L. (2013). Negative regulation of inflammation by SIRT1. Pharmacol. Res. 67, 6067.
- Gillum, M.P., Kotas, M.E., Erion, D.M., Kursawe, R., Chatterjee, P., Nead, K.T., Muise, E.S., Hsiao, J.J., Frederick, D.W., Yonemitsu, S., et al. (2011). SirT1 regulates adipose tissue inflammation. Diabetes 60, 32353245.
- Yao, H., Chung, S., Hwang, J.W., Rajendrasozhan, S., Sundar, I.K., Dean, D.A., McBurney, M.W., Guarente, L., Gu, W., Ro¨nty, M., et al. (2012). SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Invest. 122, 20322045.
- Zhang, Z., Lowry, S.F., Guarente, L., and Haimovich, B. (2010). Roles of SIRT1 in the acute and restorative phases following induction of inflammation. J. Biol. Chem. 285 , 4139141401.
- Rothgiesser, K.M., Erener, S., Waibel, S., Lu¨scher, B., and Hottiger, M.O. (2010). SIRT2 regulates NF-kB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 123, 42514258.
- Pont, A.R., Sadri, N., Hsiao, S.J., Smith, S., and Schneider, R.J. (2012). mRNA decay factor AUF1 maintains normal aging, telomere maintenance, and suppression of senescence by activation of telomerase transcription. Mol. Cell 47, 515.
- Doles, J., Storer, M., Cozzuto, L., et al., (2012). Age-associated inflammation inhibits epidermal stem cell function. Genes Dev.
- Liu, N., Landreh, M., Cao, K., Abe, M., Hendriks, G.J., Kennerdell, J.R., Zhu, Y., Wang, L.S., and Bonini, N.M. (2012). The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482, 519523.
- Shen, Y., Wollam, J., Magner, D., Karalay, O., and Antebi, A. (2012). A steroid receptor-microRNA switch regulates life span in response to signals from the gonad. Science 338, 14721476.
- Smith-Vikos, T., and Slack, F.J. (2012). MicroRNAs and their roles in aging. J. Cell Sci. 125, 717.
- Boulias, K., and Horvitz, H.R. (2012). The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF- 16/FOXO. Cell Metab. 15, 439450.
- Toledano, H., DAlterio, C., Czech, B., Levine, E., and Jones, D.L. (2012). The let-7Imp axis regulates ageing of the Drosophila testis stem-cell niche. Nature 485, 605610.
- Ugalde, A.P., Espan˜ol, Y., and Lo´pez-Ot´ın, C. (2011). Micromanaging aging with miRNAs: new messages from the nuclear envelope. Nucleus 2, 549555.
- Benetti, R., Gonzalo, S., Jaco, I., Muoz, P., Gonzalez, S., Schoeftner, S., … and Li, E. (2008). A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nature structural and molecular biology, 15(3), 268-279.
- Mitomo, S., Maesawa, C., Ogasawara, S., Iwaya, T., Shibazaki, M., et al., 2008. Downregulation of miR-138 is associated with overexpression of human telomerase reverse transcriptase protein in human anaplastic thyroid carcinoma cell lines. Cancer Sci. 99 , 280286.
- Chen, Y.F., Kao, C.H., Chen, Y.T., Wang, C.H., Wu, C.Y., et al., (2009). Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 23, 11831194.
- Lee J., Kemper, J.K.(2010) Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging, 2-8.
- Card D.A., Hebbar P.B., Li L., Trotter K.W., Komatsu Y., et al. (2008). Oct4/Sox2regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 28, 64266438.
- Hackl, M., Brunner, S., Fortschegger, K., Schreiner, C., Micutkova, L., et al. (2010). miR- 17, miR-19b, miR-20a and miR-106a are down-regulated in human aging. Aging Cell 9, 291296
- Li, G., Luna, C., Qiu, J., Epstein, D.L., Gonzalez, P. (2009). Alterations in miRNA expression in stress-induced cellular senescence. Mech. Ageing Dev. 130, 731741.
- Fontana, L., Fiori, M.E., Albini, S., Cifaldi, L., Giovinazzi, S., et al. (2008). Antagomir17- 5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One 3, e2236.
- Taganov, K.D., Boldin, M.P., Chang, K.J., Baltimore, D., 2006. NF-κB-dependent induction of miRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103, 12481 12486.
- Iliopoulos, D., Hirsch, H.A., Struhl, K. (2009). An epigenetic switch involving NFκB, Lin28, Let-7 MiRNA, and IL6 links inflammation to cell transformation. Cell 139 , 693706
- Quinn, S. R., O’Neill, L. A. (2011). A trio of microRNAs that control Toll-like receptor signalling. International immunology, 23(7), 421-425.
- Olivieri, F., Rippo, M. R., Procopio, A. D., Fazioli, F. (2014). Circulating inflammamiRs in aging and age-related diseases. The origin, function and diagnostic potential of extracellular microRNA in human body fluids, 106.
- Boldin, M. P., Baltimore, D. (2012). MicroRNAs, new effectors and regulators of NFB. Immunological reviews, 246(1), 205-220.
- Steinmann, G. G., Klaus, B., Mu¨llerHermelink, H. K. (1985). The involution of the ageing human thymic epithelium is independent of puberty. Scandinavian journal of immunology, 22(5), 563-575.
- Steinmann, G. G. (1986). Changes in the human thymus during aging. In The Human Thymus (pp. 43-88). Springer Berlin Heidelberg.
- Lynch, H. E., Goldberg, G. L., Chidgey, A., Van den Brink, M. R., Boyd, R., Sempowski, G. D. (2009). Thymic involution and immune reconstitution. Trends in immunology, 30(7), 366-373.
- Weksler, M. E., Pawelec, G., Franceschi, C. (2009). Immune therapy for age-related diseases. Trends in immunology, 30(7), 344-350.
- Chen, W. H., Kozlovsky, B. F., Effros, R. B., Grubeck-Loebenstein, B., Edelman, R., Sztein, M. B. (2009). Vaccination in the elderly: an immunological perspective. Trends in immunology, 30(7), 351-359.
- Dowling, M. R., Hodgkin, P. D. (2009). Why does the thymus involute? A selectionbased hypothesis. Trends in immunology, 30(7), 295-300.
- Swaab, D. F., Hofman, M. A., Lucassen, P. J., Purba, J. S., Raadsheer, F. C., Van de Nes, J. A. P. (1993). Functional neuroanatomy and neuropathology of the human hypothalamus. Anatomy and embryology, 187(4), 317-330.
- Bartke, A. (2011). Pleiotropic effects of growth hormone signaling in aging. Trends Endocrinol. Metab. 22, 437442.
- Zhang, G., Li, J., Purkayastha, S., Tang, Y., Zhang, H., Yin, Y., Li, B., Liu, G., and Cai, D. (2013). Hypothalamic programming of systemic ageing involving IKK-b, NF-kB and GnRH. Nature 497, 211216.
- Satoh, A., Brace, C. S., Rensing, N., Cliften, P., Wozniak, D. F., Herzog, E. D., … Imai, S. I. (2013). Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell metabolism, 18(3), 416-430.
- Imai, S. I., Guarente, L. (2014). NAD+ and sirtuins in aging and disease. Trends in cell biology, 24(8), 464-471.
- Chang, H., Guarente, L. (2013). SIRT1 and other sirtuins in metabolism Trends in Endocrinology and Metabolism, 18.
- Kumar, S., Lombard, D. B. (2015). Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxidants and redox signaling, 22(12), 1060-1077.
- Rodier, F., Campisi, J., Bhaumik, D. (2007). Two faces of p53: aging and tumor suppression. Nucleic acids research, 35(22), 7475-7484.
- Maiuri, M.C., Galluzzi L., Morselli E., Kepp O., Malik S.A., Kroemer G. (2010). Autophagy regulation by p53 Current Opinion in Cell Biology, 22:181185
- Kroemer, G., Marin˜o, G., Levine, B. (2010). Autophagy and the Integrated Stress Response. Molecular Cell 40.
- Matoba, S., Kang, J.G., Patino, W.D., Wragg, A., Boehm, M., Gavrilova, O., Hurley, P.J., Bunz, F., Hwang, P.M. (2006). p53 Regulates Mitochondrial Respiration. Science 312, 1650.
- Ferbeyre, G., Lowe, S. W. (2002). Ageing: The price of tumour suppression?. Nature, 415(6867), 26-27.
- Morimoto, R.I., Cuervo, A.M. (2014). Proteostasis and the aging proteome in health and disease. J. Gerontol. A Biol. Sci. Med. Sci. 69 (suppl. 1), S33S38
- Verdin, E., Hirschey, M. D., Finley, L. W., Haigis, M. C. (2010). Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends in biochemical sciences, 35(12), 669-675.
- Palikaras, K., Tavernarakis, N. (2012). Mitophagy in neurodegeneration and aging. Frontiers in genetics, 3, 297.
- Canto, C., Auwerx, J. (2009). PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current opinion in lipidology, 20(2), 98.
- Shih, J., Donmez, G. (2013). Mitochondrial sirtuins as therapeutic targets for agerelated disorders. Genes and cancer, 4(3-4), 91-96.
- Fernandez-Marcos, P.J., and Auwerx, J. (2011). Regulation of PGC-1a, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884S890 S.
- Iyer, N. V., Kotch, L. E., Agani, F., Leung, S. W., Laughner, E., Wenger, R. H., … Semenza, G. L. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1. Genes and development, 12(2), 149-162.
- Semenza, G. L. (2003). Targeting HIF-1 for cancer therapy. Nature reviews cancer, 3(10), 721-732.
- Haigis, M. C., Guarente, L. P. (2006). Mammalian sirtuinsemerging roles in physiology, aging, and calorie restriction. Genes and development, 20(21), 2913-2921.
- Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., … Sinclair, D. A. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science, 305(5682), 390-392.








